Erythropoietin after focal cerebral ischemia activates the janus kinase–signal transducer and activator of transcription signaling pathway and improves brain injury in postnatal day 7 rats
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Erythropoietin (Epo) plays a central role in erythropoiesis but also has neuroprotective properties. Recently, Epo-related neuroprotective studies used a hypoxic-ischemic neonatal
model, which is different from focal stroke, a frequent cause of neonatal brain injury. We report on the effects of Epo treatment given after focal stroke and its potential neuroprotective
mechanisms in postnatal day 7 rats with focal cerebral ischemia (FCI) achieved by occlusion of the middle cerebral artery. The experimental groups included sham operation, FCI plus vehicle,
and FCI plus Epo. In the Epo-treated group, pups received a single intraperitoneal injection of 1000 U/kg 15 min after FCI or three injections of 100, 1000, or 5000 U/kg, starting at 15 min
and repeated at 1 and 2 d after FCI. Epo treatment produced significant reductions in the mean infarct area and volume at 1 and 3 d after FCI, demonstrated by 2,3,5-triphenyltetrazolium
chloride staining. Terminal deoxynucleotidyltransferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL) staining showed a markedly reduced number of TUNEL-positive
cells in the Epo-treated group when compared with the vehicle control 3 d after FCI (_p_ < 0.01). The most effective dose after FCI was 1000 U/kg for 3 d. Immunoanalyses showed that Epo
induced a significant increase in phosphorylated Janus kinase 2 and signal transducer and activator of transcription-5 expressions at 1 and 3 d and up-regulated Bcl-xL expression by 24 h
after FCI but did not affect Epo receptor or NF-κB expression. In conclusion, Epo given after FCI in neonatal rats provides significant neuroprotection, mediated possibly by activation of
the Janus kinase–signal transducer and activator of transcription–Bcl-xL signaling pathways. SIMILAR CONTENT BEING VIEWED BY OTHERS COMPLEXIN 2 CONTRIBUTES TO THE PROTECTIVE EFFECT OF NAD+
ON NEURONAL SURVIVAL FOLLOWING NEONATAL HYPOXIA-ISCHEMIA Article 17 April 2025 CELASTROL AMELIORATES HYPOXIC-ISCHEMIC BRAIN INJURY IN NEONATAL RATS BY REDUCING OXIDATIVE STRESS AND
INFLAMMATION Article Open access 20 May 2024 NRF2 ACTIVATION AMELIORATES BLOOD–BRAIN BARRIER INJURY AFTER CEREBRAL ISCHEMIC STROKE BY REGULATING FERROPTOSIS AND INFLAMMATION Article Open
access 04 March 2024 MAIN Erythropoietin (Epo), a glycoproteic hormone produced by the kidney, not only affects the hematopoietic system but also exerts a multifunctional trophic influence
on the general homeostasis of the entire organism (1,2). The recent discovery of a specific Epo receptor (EpoR) in the CNS (1,3) has led to investigations in the neurotrophic and
neuroprotective effects of Epo on the developing brain. _In vitro_ studies have shown that Epo protects primary cultured neurons against injuries induced by glutamate deprivation, hypoxia,
glucose, kainic acid, or serum (4–8). Moreover, _in vivo_ studies demonstrate that Epo attenuates cerebral ischemic damage in adult gerbils, rats, and mice (4,6,8–10). In neonatal rat
models, Epo prevents intrauterine ischemia-reperfusion– or _N_-methyl-d-aspartate receptor antagonist–induced brain injury (11,12) and reduces brain injury in a hypoxic-ischemic model of the
postnatal day 7 (P7) rat (13,14). The latter neonatal rat model requires both unilateral carotid artery ligation and systemic hypoxia (15,16) but does not accurately reproduce the
pathogenesis of focal stroke in human neonates. Recent epidemiologic studies suggest that the incidence of neonatal focal cerebral ischemia (FCI) is higher than previously recognized (17–19)
and in fact may be more prevalent than global cerebral ischemia from systemic asphyxia (18). Therefore, it is critically important to evaluate the effects of Epo on focal brain injury by
using an appropriate neonatal FCI model. Epo and its receptor have been shown to play a crucial role in neuronal survival and recovery (1,3). Numerous pieces of evidence indicate that Epo
and its receptor are expressed in both neonatal and adult brains (1–3) and are up-regulated by hypoxia and ischemia (6), and Epo concentration is significantly increased in cerebrospinal
fluid of asphyxiated human neonates (20). Recently, using a modified permanent FCI model in the P7 rat (21), we found that FCI induces a significant increase in EpoR expression in the
neonatal rat brain, and EpoR was expressed on neurons, microglia/macrophage, and endothelium of blood vessels in the ischemic hemisphere 24 h after FCI (22). Furthermore, terminal
deoxynucleotidyltransferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL) staining indicated that nearly half of EpoR-positive cells were also TUNEL positive in
the ischemic cortex 24 h after FCI (22). Although TUNEL staining is not a specific test for detecting cell apoptosis and a supplemental test such as detecting caspase activation is required
to confirm whether TUNEL-positive cells undergo apoptosis, our results suggest that EpoR-positive cells may undergo apoptosis-like cell death in ischemic areas. Given these findings, we
theorize that endogenous Epo is produced by certain populations of cells in the brain and that under normal conditions, neuronal survival in the developing brain is maintained, at least in
part, by endogenous Epo. We further speculate that with significant ischemic stress to the brain, the endogenous Epo pool may be insufficient for neuronal homeostasis and protection and that
the relative insufficiency of Epo during such periods of ischemic stress then may trigger neuronal apoptosis. Therefore, we hypothesized that provision of exogenous Epo after FCI mitigates
FCI-induced brain injury. It is well known that in nonneuronal cells, the effects of Epo are mediated by its receptor, which subsequently activates Janus kinase 2 (Jak2), leading to tyrosine
phosphorylation of the signal transducer and activator of transcription 5 (Stat5) and the up-regulation of antiapoptotic genes (23,24). Recent data have shown that Jak2-Stat5 were expressed
in the various brain regions during embryonic and postnatal stages, suggesting that Jak2-Stat5 may be involved in brain development (23). In cultured neuronal cells, Epo has been
demonstrated to protect cerebrocortical neurons from excitotoxic- and nitric oxide–induced damage by triggering cross-talk between Jak2 and nuclear factor-κB (NF-κB) signaling pathways (25).
NF-κB has been shown to up-regulate transcription of other antiapoptotic proteins such as inhibitors of apoptosis protein (XIAP and c-IAP), which block activation of specific cell-death
caspases and subsequent apoptosis (26). Moreover, Epo has further been found to significantly up-regulate expression of pro-survival Bcl-xL mRNA and protein in the ischemic hippocampal CA1
field of adult gerbils, compared with vehicle treatment (10). Bcl-xL has been shown to suppress apoptosis in part by blocking the release of cytochrome c from mitochondria, which is a
critical step in the activation of the caspase protease cascade (27). However, it is not known whether these events also occur in neonatal brains with FCI. Given this background evidence and
our hypothesis, our study aims were to investigate the effects of Epo on brain injury induced by FCI in neonatal rats and some of the potential intracellular mechanisms by which Epo may
prevent neonatal stroke-induced brain injury, specifically evaluating the effects of Epo on the expression of EpoR, phosphorylated Jak2 (p-Jak2) and Stat5 (p-Stat5), and the antiapoptotic
genes Bcl-xL and NF-κB in neonatal rat brains after focal ischemic insult. METHODS ANIMALS. Timed pregnant Sprague-Dawley rats that were carrying 18-d-old fetuses (E18) were purchased from
Charles River Laboratories (Wilmington, MA). Pregnant dams were housed in a temperature- and light-controlled animal care facility and given food and water _ad libitum_. All animal research
was approved by the Emory University Institutional Animal Care Committee and performed in accordance with National Institutes of Health animal care guidelines. FCI MODEL. To produce
permanent FCI, we used a modified intraluminal catheter technique to occlude the middle cerebral artery (MCA) in the P7 rat pups, as we have previously described (21). Briefly, each pup was
weighed, anesthetized with 2% isofluorane, and immobilized in a supine position. Under an operative microscope, the left common carotid artery (CCA), the external carotid artery (ECA), and
the internal carotid artery (ICA) were separated from adjacent veins and nerves. After ligation of the CCA and ECA, a suture embolus distally coated with silicone resin (using a size
appropriate for the pup's body weight) was inserted through a small incision on the CCA just proximal to the CCA-ECA-ICA junction. The suture embolus then was advanced 7–8 mm so that
its tip effectively occluded the MCA. During the operation, the pup's body temperature was monitored continuously using a rectal temperature probe, and euthermia was maintained with the
assistance of an overhead heating lamp. During recovery from anesthesia, the pups were kept in a chamber at room air with an environmental temperature of 37°C for 15 min and then returned
to their dams. Sham operation involved the same ventral neck incision with dissection of the pertinent vessels but without insertion of a suture embolus. EXPERIMENTAL GROUPS AND TREATMENT
WITH EPO AND VEHICLE. The 140 pups for this study were divided into three groups: sham-operated plus vehicle (_n_ = 30), FCI plus vehicle (_n_ = 45), and FCI plus Epo (_n_ = 65). Recombinant
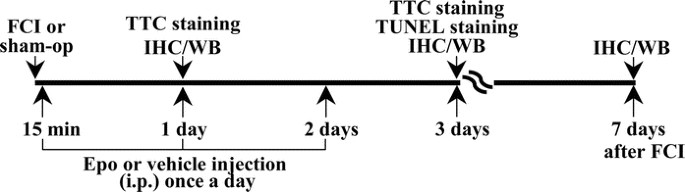
human Epo (4000 U/mL; Amgen Inc., Thousand Oaks, CA) was diluted in a vehicle of 0.01 M PBS (pH 7.4) and 0.1% BSA to a final concentration of 80 U/mL. The experimental schedule is shown in
Fig. 1. For the studies performed 1 d after FCI, the pups received a single intraperitoneal injection of either Epo at 1000 U/kg or vehicle 15 min after FCI (_n_ = 13 pups in each group).
For the analyses and studies performed at 3 and 7 d after FCI, 52 different pups received their first Epo injection 15 min after FCI and then two additional injections once a day for 2 d at
100, 1000, or 5000 U/kg each dose. A comparison FCI placebo control group (_n_ = 39) received injections of the vehicle at the same volume and schedule as the Epo group. The sham-operated
group received only vehicle injection as normal control. The Epo dose and the schedule of administration were chosen according to previous protocols that evaluated the drug in a
hypoxic-ischemic model in P7 rat pups (13,14,28). The treatment groups were randomly assigned, and the investigators who evaluated outcomes were blinded to the treatment received.
MEASUREMENT OF INFARCT AREA AND VOLUME. For determining the infarct area and volume, 2,3,5-triphenyltetrazolium chloride (TTC) staining was performed after the rat pups were anesthetized
with pentobarbital and decapitated at 1 d (_n_ = 5 for 1000 U/kg of Epo- or vehicle-treated FCI group) and at 3 d (_n_ = 10 for 100, 1000, or 5000 U/kg of Epo- or vehicle-treated FCI group)
after FCI. Their brains were harvested and coronally sectioned into five 2-mm slices, which then were incubated in a 1% TTC solution at 37°C for 20 min and fixed in 10% buffered formalin at
4°C overnight. An investigator who was blinded to the treatment group obtained photographs of the sections with a digital camera and measured the infarct area using Windows Image J (similar
to NIH Image). The total infarct volume was calculated as the sum of the infarct areas from the 2-mm-thick continuous coronal sections. _IN SITU_ DETECTION OF DNA FRAGMENTATION. For
estimating the number of degenerating cells 3 d after FCI, TUNEL staining was conducted using an ApopTag peroxidase _in situ_ apoptosis detection kit (Serologicals, Norcross, GA). The
sham-operated (_n_ = 6) and FCI (_n_ = 6 for 1000 U/kg of Epo- or vehicle-treated animals) pups were anesthetized with pentobarbital and transcardially perfused with 4% paraformaldehyde 3 d
after operation. Four serial coronal sections, including the dorsal hippocampus (1.0–1.12 mm posterior to the bregma) were processed for TUNEL staining (29). Briefly, the sections were
incubated _1_) in a mixture of terminal deoxynucleotidyltransferase and reaction buffer that contained digoxigenin-dUTP-biotin in a humidified chamber for 1 h at 37°C, _2_) incubated with
anti-digoxigenin peroxidase for 1 h at room temperature, and _3_) exposed to 0.05% diaminobenzidine and 0.02% hydrogen peroxide. IMMUNOHISTOCHEMISTRY ANALYSIS OF EPOR. The sham-operated or
FCI animals with or without Epo (1000 U/kg) injection were anesthetized with pentobarbital and perfused transcardially with 4% paraformaldehyde at 1, 3, and 7 d after operation (_n_ = 4 for
each time point in each group). The brains were excised and immersed overnight in 0.1 M phosphate buffer (pH 7.4) that contained 30% sucrose at 4°C. Four serial coronal sections 30 μm thick
at the same level of the dorsal hippocampus for TUNEL staining as described above were cut with a cryostat and processed for immunohistochemistry (IHC) with an affinity-purified rabbit
antibody against EpoR (Santa Cruz Biotechnology, Santa Cruz, CA). In brief, the sections were _1_) washed with 0.1 M PBS for 30 min; _2_) incubated in 3% hydrogen peroxide for 10 min to
quench endogenous peroxidase; _3_) incubated in blocking solution that contained 10% normal goat serum, 0.3% Triton X-100, and 0.1% BSA in PBS for 1 h at room temperature; _4_) incubated
with the primary antibody (1:100) diluted with PBS that contained 1% normal goat serum and 0.3% Triton X-100 for 48 h at 4°C; and _5_) incubated with FITC-conjugated anti-rabbit IgG (Sigma
Chemical Co., St. Louis, MO) for 2 h. Control immunoreactions excluded the incubation of the first antibody. COUNTING TUNEL- OR EPOR-POSITIVE CELLS. An investigator who was blinded to the
treatment group counted TUNEL- or EpoR-positive cells in the left cortex of the sham-operated or FCI animals with or without Epo treatment in the four serial coronal sections. The mean
number of positive cells was calculated for each section in each animal. WESTERN BLOT ANALYSIS OF EPOR, P-JAK2, P-STAT5, BCL-XL, OR NF-ΚB. Homogenates of the cortices were obtained from the
sham-operated and FCI animals with or without Epo (1000 U/kg) injection at 1, 3, and 7 d after operation (_n_ = 4 for each time point in each group). They were solubilized in a sample
solution that contained 2 mM EGTA, 0.01 mM phenylmethane sulfonyl fluoride, and 2% SDS. An equal amount of protein (40 μg) in the homogenates was electrophoresed in individual lanes using
10% polyacrylamide gel in the Laemmli's buffer system (30). Protein concentration was determined by BCA protein assay reagent (Pierce Chemical Co., Rockford, IL) with BSA as a standard.
The electrophoretic bands were transferred to nitrocellulose sheets (31) and immunoblotted with an antibody of EpoR (1:100), p-Jak2 (1:200), p-Stat5 (1:200), Bcl-xL (1:200), or NF-κB
(1:100). These primary antibodies were purchased from Santa Cruz Biotechnology. Anti-rabbit IgG coupled with alkaline phosphatase (Promega, Madison, WI) was used for the second
immunoreaction. The immunoreactive bands were visualized with nitrotetrazolium blue chloride and 5-bromo-4-chloro-3-indolyl phosphate as described elsewhere (32). Prestained molecular weight
markers were purchased from Bio-Rad Laboratories (Richmond, CA) and used to monitor the separation of proteins while electrophoresis was in progress. The Western blot analyses were repeated
four times. For quantitative evaluation, the immunoreactive bands were subjected to densitometric analysis with a combination of Adobe Photoshop and the Windows Image (29,33). STATISTICAL
ANALYSIS. The data of each group are summarized as their mean ± SD. The two-tailed Mann-Whitney _U_ test was used to evaluate differences in mean infarct area, volume, and numbers of
TUNEL-positive cells between Epo- and vehicle-treated pups. Statistical analyses for the data of IHC and Western blot were conducted using ANOVA followed by Fisher's post hoc test.
Statistical significance was set at 0.05. RESULTS SURVIVAL AND BODY WEIGHT. All pups that underwent either sham operation or FCI survived to the end of the study period. However, pups that
underwent FCI had a body weight that was significantly lower than that of the sham-operated pups 3 d after FCI (Epo-treated group: 16.5 ± 0.8 g; vehicle-treated FCI group: 14.0 ± 1.8 g, _p_
= 0.8; sham-operated group: 20.0 ± 1.0 g; _p_ < 0.05). EFFECTS OF EPO ON INFARCT AREA AND VOLUME. TTC staining revealed a reproducible infarct area in the hemisphere ipsilateral to the
MCA occlusion in vehicle-treated FCI pups at 1 and 3 d after FCI. The infarct area was demonstrated by a lack of TTC staining and a “pale-white” color (Fig. 2_A_). In comparison, TTC
staining of the contralateral hemisphere showed no evidence of infarction or appreciable injury in vehicle- or Epo-treated FCI pups, demonstrated by a deep red stain of cortical tissue (Fig.
2_A_ and _B_). One dose of Epo at 1000 U/kg after FCI significantly reduced the ipsilateral infarct area at the levels irrigated by the MCA by 1 d after FCI (Fig. 3_A_). A significant
change was also observed 3 d after FCI, with three doses of Epo (Figs. 2_B_ and 4_A_). Epo treatment also reduced significantly the infarct volume (Figs. 3_B_ and 4_B_) compared with the
vehicle-treated FCI group. Striking, 3 d after FCI, there was no evidence of infarction in the brains of three of 10 pups that were treated with three doses of Epo (1000 U/kg) after FCI
(Fig. 2_B_). The mean infarct area and volume in the vehicle-treated FCI group 3 d after FCI were 64.8 ± 3.2 and 129.6 ± 6.5 mm3, respectively (Fig. 4_A_ and _B_). Epo treatment
significantly reduced the mean infarct area and volume 3 d after FCI in a dose-dependent manner (Fig. 4). The most effective Epo dose in this experimental protocol was 1000 U/kg, causing a
40% reduction in the mean infarct area (24.5 ± 5.4 mm2; U = 0.0, _p_ < 0.001) and in volume (48.9 ± 10.8 mm3; U = 0.0, _p_ < 0.001) 3 d after FCI when compared with vehicle-treated FCI
pups (Fig. 4_A_ and _B_). EFFECTS OF EPO ON THE NUMBER OF TUNEL-POSITIVE CELLS IN THE ISCHEMIC CORTEX. Few TUNEL-positive cells were detected in the cortex of sham-operated animals (Fig.
5_A_), with a mean number of TUNEL-positive cells of 53.0 ± 13.8 cells/section (Fig. 5_D_). FCI induced a significant increase in the number of TUNEL-positive cells in the cortex of the
vehicle-treated FCI group (Fig. 5_B_ and _D_). Epo treatment after FCI significantly attenuated this (Fig. 5_C_ and _D_). Among the pups that underwent FCI, the number of TUNEL-positive
cells in the cortex of vehicle-treated rats increased nearly 10-fold (511.9 ± 39.2 cells/section) 3 d after FCI (Fig. 5_B_ and _D_), compared with the sham-operated pups (U = 0.0, _p_ <
0.001). Epo treatment after FCI (three doses at 1000 U/kg each) significantly reduced the number of TUNEL-positive cells (132.7 ± 24.3 cells/sections) in the ischemic cortex compared with
the vehicle-treated FCI pups (U = 1.0, _p_ < 0.05; Fig. 5_C_ and _D_). EFFECTS OF EPO TREATMENT ON EPOR EXPRESSION IN THE ISCHEMIC CORTEX. Assuming that intraperitoneally administered Epo
penetrates to the ischemic hemisphere to rescue ischemic cells _via_ activation of local EpoR, we investigated whether Epo treatment altered EpoR expression in the ischemic cortex from 1 to
7 d after FCI. IHC analysis 1 d after FCI showed that FCI caused a significant increase in the number of EpoR-positive cells in the ischemic cortex of FCI rats that were treated with
vehicle (Fig. 6_A_ and _H_), compared with the sham-operated animals (Fig. 6_G_ and _H_; *_p_ < 0.01, tested by ANOVA followed by Fisher's _post hoc_ test). This was consistent with
our previously reported findings (21). Thereafter, at 3 and 7 d after FCI, this enhanced EpoR immunoreactivity declined to the level of sham-operated animals (Fig. 6_C_, _E_, and _H_). Epo
treatment had no significant influence on EpoR expression in the ischemic cortex (_p_ > 0.05; Fig. 6_H_). The number of EpoR-positive cells was similar in the Epo-treated group (1000
U/kg; Fig. 6_B_, _D_, _F_, and _H_) and in the vehicle-treated FCI group (Fig. 6_A_, _C_, _E_, and _H_) from 1 to 7 d after FCI. These findings were verified by subsequent Western blot
analysis (Figs. 7 and 8_A_). EFFECTS OF EPO TREATMENT ON P-JAK2, P-STAT5, BCL-XL, OR NF-ΚB EXPRESSION IN THE ISCHEMIC CORTEX. Western blot analysis showed a distinct constitutive expression
of EpoR, p-Jak2, p-Stat5, Bcl-xL, or NF-κB in the cortex of sham-operated rats (Fig. 7, lane 1). In the vehicle-treated FCI group, FCI caused significant increases in EpoR expressions at 1 d
(Fig. 7 lane 2; Fig. 8_A_) and also in Bcl-xL expression at 3 d (Fig. 7, lane 4; Fig. 8_D_) but caused marked declines in p-Jak2 and p-Stat5 expressions from 1 to 7 d after FCI (Fig. 7,
lanes 2, 4, and 6 of p-Jak2 and p-Stat5; Fig. 8_B_ and _C_). When compared with the vehicle-treated FCI group, Epo treatment (1000 U/kg) did not significantly affect EpoR expression from 1
to 7 d (Fig. 7, lanes 3, 5, 7; Fig. 8_A_) but prevented the decrease in p-Jak2 or p-Stat5 expression at 1 and 3 d (Fig. 7, lanes 3, 5; Fig. 8_B_ and _C_). Moreover, it caused a marked
increase in Bcl-xL expression 1 d (Fig. 7, lane 3; Fig. 8_D_) after FCI (_p_ < 0.05; ANOVA followed by Fisher's _post hoc_ test). However, NF-κB expression in the cortex was not
altered by FCI or Epo treatment from 1 to 7 d after FCI (Fig. 7, lanes 2–7; Fig. 8_E_). These results suggest that exogenous Epo administration did not alter the FCI-induced transient
up-regulation of EpoR expression in the ischemic cortex from 1 to 7 d after FCI; however, Epo activated p-Jak2, p-Stat5, and Bcl-xL at 1 and 3 d after FCI. DISCUSSION Numerous studies in
adult animals demonstrate that treatment with Epo attenuates brain injury in hypoxia, transient forebrain ischemia, or FCI (5,6,9–10). In neonatal animals, Epo has been shown to reduce
neurotoxicity of _N_-methyl-d-aspartate receptor antagonists (12) and prevent brain injury in a P7 hypoxic-ischemic rat model (13,14,28) when the first dose is given before producing the
injury. There are two important differences between our current study and previous ones. One important difference is that in this study, Epo was applied after FCI, whereas previous studies
involved Epo pretreatment before the insult. The other important difference is that the cerebral insult in the hypoxic-ischemic model is different from that in our FCI model. The current FCI
model induces a focal ischemic stroke in the region supplied by the MCA and is relevant for future studies given that neonatal focal cerebral insults are more common than previously
recognized (34,35). The present study in this FCI model in P7 rat pups demonstrates that Epo administered exogenously after the injury significantly reduces the mean infarct area, the mean
infarct volume, and the number of TUNEL-positive cells in the ipsilateral ischemic cortex in a dose-dependent manner. Therefore, this study provides more practical meaning to Epo treatment
for stroke in human neonates. In the current experiments, we focused only on the effects of Epo on infarct size at 1 and 3 d after FCI by using TTC staining, which is an optimal method for
identification of cerebral infarct (36). However, infarct size has been shown to evolve continually after 3 d of brain insult in many stroke models (37). Therefore, it is not clear whether
the neuroprotective effects of Epo at 1 and 3 d after FCI are caused by reducing infarct size, inhibiting infarct expansion, or both. A recent report has shown that Epo administration at a
single dose immediately after the hypoxia period significantly improves long-term spatial memory deficits at 20 wk and reduces the infarct volume at 21 wk after hypoxia-ischemia in P7 rats
(38). More important, the brains in the Epo-treated animals show a slight hypotrophy of the ischemic hemisphere, whereas the brains in the saline-treated animals exhibit a severe atrophy and
cortical cavities at 21 wk after hypoxia-ischemia (38). In addition, our ongoing experiments indicate that Epo causes a significant reduction in the infarct area and volume at 6 and 12 wk
after FCI (unpublished data). These results suggest that exogenous Epo treatment has a long-term neuroprotection on the developing brain against injury and that its effects may be caused not
only by delaying infarct expansion but also by reducing infarct size. The body size of rats that underwent FCI surgery was significantly decreased compared with sham-operated rats. Ligation
of the ECA is necessary in this FCI model and may have affected swallowing functions, resulting in less weight gain during the first postsurgical week in all pups that underwent FCI. A
similar finding has been reported in adult rats with FCI (39). However, it is unlikely that this is a significant confounder to the significant differences in outcomes noted between Epo- and
vehicle-treated rats, both of which underwent the FCI surgery and had similar decreases in weight gain compared with sham-operated rats. Nonetheless, this is a weakness of the model when
comparing outcomes with sham-operated subjects. Endogenous Epo may act as a neuroprotectant in the brain (1,3). Indeed, Sakanaka _et al._ (5) demonstrated that intraventricular infusion of a
soluble EpoR, which can neutralize endogenous Epo, caused neuronal death in the hippocampus of gerbils that were subjected to a mild ischemia that did not result in neuronal damage by
itself. This result was confirmed by an observation that neutralization of endogenous Epo exacerbated neuronal injury in a transient global retina ischemic model (40). Furthermore, Shingo
_et al._ (41) demonstrated that hypoxia-induced endogenous Epo may promote the production of neuronal progenitors after hypoxia. In the current experiments, we found that exogenously
administered Epo after focal ischemic insult significantly reduced the infarct area, the infarct volume, and the number of TUNEL-positive cells in the ischemic cortex. These results suggest
that endogenous Epo may be a protective agent for brains against cerebral ischemia _in vivo_. The important issue to be addressed is whether the neuroprotective effects of Epo are initially
triggered by its binding to its receptor. We did not detect any significant alteration in EpoR expression in the ischemic cortex from 1 to 7 d after focal ischemic insult in the Epo-treated
pups compared with vehicle-treated pups. This issue is consistent with the findings that Epo treatment does not alter EpoR mRNA levels in a neurotoxic model (12) and that Epo application for
3 d does not down-regulate EpoR in a neonatal hypoxic-ischemic model (28). Recent data have also shown that carbamylated Epo or certain Epo mutants do not bind to the classical EpoR, but
these Epo mutants were neuroprotective _in vitro_ and _in vivo_ (42). Therefore, the neuroprotective effect of exogenous Epo administration may not down-regulate the ischemia-induced
up-regulation of EpoR but seems to restore endogenous Epo signaling. However, further experiments are needed to prove whether Epo binds to its receptor within the first 24 h after FCI and
hence prevents the need for up-regulation of EpoR in the subsequent days after the initial 24 h of FCI and exogenous Epo administration. The precise molecular mechanism underlying the
neuroprotective effects of Epo has not been completely elucidated. Epo may act at multiple levels, including its antiapoptotic mechanisms. In nonneuronal cells, numerous pieces of evidence
indicate that the antiapoptotic effects of Epo are mediated by activating Jak2, leading to tyrosine phosphorylation of the Stat5 and the up-regulation of antiapoptotic genes (23,24).
Recently, this Epo-mediated molecular signaling pathway was also established in neurons. Epo has been shown to protect hippocampal neurons from ischemic neuronal damage through activation of
Jak2 (43) and induce phosphorylation of Stat5 in rat hippocampal neurons (44). In addition, Campana and Myers (45) reported that systemic administration of Epo reduces dorsal root ganglion
apoptosis by activating p-Jak2 within 1 d after L5 spinal nerve crush. Furthermore, Digicaylioglu and Lipton (25) demonstrated that Epo prevents cultured cerebrocortical neuronal death by
triggering Jak2 signaling pathways. More important, the current experiments indicated that Epo significantly activates p-Jak2 and p-Stat5 at 1 and 3 d and causes a significant increase in
downstream cellular neuroprotective gene Bcl-xL 1 d after FCI in the neonatal brains. Epo treatment did not alter the expression of NF-κB, which has been shown to be activated by Epo
treatment in cultured cerebrocortical neurons (25). However, the present results cannot exclude a possibility that Epo treatment may activate NF-κB's transcriptional activating
function. This evidence suggests that Epo enhances neuronal survival possibly through activation of Jak2-Stat5 and the subsequent downstream cellular neuroprotective genes such as Bcl-xL. In
conclusion, the findings of this study show that Epo given after FCI protects the neonatal brain against FCI insults and attenuates the effects of neonatal cerebral stroke in neonatal rats
with FCI, providing more practical meaning to Epo treatment for stroke in human neonates. Furthermore, the findings that exogenous Epo activated p-Jak2, p-Stat5, and Bcl-xL at 1 and 3 d
after FCI provide insight regarding the molecular mechanisms involved in Epo neuroprotection in the developing brain. ABBREVIATIONS * CCA: common carotid artery * ECA: external carotid
artery * Epo: erythropoietin * EpoR: Epo receptor * FCI: focal cerebral ischemia * ICA: internal carotid artery * IHC: immunohistochemistry * Jak2: Janus kinase-2 * MCA: middle cerebral
artery * Stat5: signal transducer and activator of transcription-5 * TTC: 2,3,5-triphenyltetrazolium chloride; * TUNEL: terminal deoxynucleotidyltransferase-mediated 2′-deoxyuridine
5′-triphosphate-biotin nick end labeling REFERENCES * Dame C, Juul SE, Christensen RD 2001 The biology of erythropoietin in the central nervous system and its neurotrophic and
neuroprotective potential. _Biol Neonate_ 79: 228–235 Article CAS PubMed Google Scholar * Buemi M, Cavallaro E, Floccari F, Sturiale A, Aloisi C, Trimarchi M, Corica F, Frisina N 2003
The pleiotropic effects of erythropoietin in the central nervous system. _J Neuropathol Exp Neurol_ 62: 228–236 Article CAS PubMed Google Scholar * Fisher JW 2003 Erythropoietin:
physiology and pharmacology update. _Exp Biol Med_ 228: 1–14 Article CAS Google Scholar * Morishita E, Masuda S, Nagao M, Yasuda Y, Sasaki R 1997 Erythropoietin receptor is expressed in
rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. _Neuroscience_ 76: 105–116 Article CAS PubMed Google Scholar *
Sakanaka M, Wen TC, Matsuda S, Masuda S, Morishita E, Nagao M, Sasaki R 1998 In vivo evidence that erythropoietin protects neurons from ischemic damage. _Proc Natl Acad Sci USA_ 95:
4635–4640 Article CAS PubMed PubMed Central Google Scholar * Bernaudin M, Marti HH, Roussel S, Divoux D, Nouvelot A, MacKenzie ET, Petit E 1999 A potential role for erythropoietin in
focal permanent cerebral ischemia in mice. _J Cereb Blood Flow Metab_ 19: 643–651 Article CAS PubMed Google Scholar * Sinor AD, Greenberg DA 2000 Erythropoietin protects cultured
cortical neurons, but not astroglia, from hypoxia and AMPA toxicity. _Neurosci Lett_ 290: 213–215 Article CAS PubMed Google Scholar * Siren AL, Fratelli M, Brines M, Goemans C,
Casagrande S, Lewczuk P, Keenan S, Gleiter C, Pasquali C, Capobianco A, Mennini T, Heumann R, Cerami A, Ehrenreich H, Ghezzi P 2001 Erythropoietin prevents neuronal apoptosis after cerebral
ischemia and metabolic stress. _Proc Natl Acad Sci USA_ 98: 4044–4049 Article CAS PubMed PubMed Central Google Scholar * Brines ML, Ghezzi P, Keenan S, Agnello D, de Lanerolle NC,
Cerami C, Itri LM, Cerami A 2000 Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. _Proc Natl Acad Sci USA_ 97: 10526–10531 Article CAS PubMed
PubMed Central Google Scholar * Wen TC, Sadamoto Y, Tanaka J, Zhu PX, Nakata K, Ma YJ, Hata R, Sakanaka M 2002 Erythropoietin protects neurons against chemical hypoxia and cerebral
ischemic injury by up-regulating Bcl-xL expression. _J Neurosci Res_ 67: 795–803 Article CAS PubMed Google Scholar * Solaroglu I, Solaroglu A, Kaptanoglu E, Dede S, Haberal A, Beskonakli
E, Kilinc K 2003 Erythropoietin prevents ischemia-reperfusion from inducing oxidative damage in fetal rat brain. _Childs Nerv Syst_ 19: 19–22 PubMed Google Scholar * Dzietko M,
Felderhoff-Mueser U, Sifringer M, Krutz B, Bittigau P, Thor F, Heumann R, Buhrer C, Ikonomidou C, Hansen HH 2004 Erythropoietin protects the developing brain against N-methyl-D-aspartate
receptor antagonist neurotoxicity. _Neurobiol Dis_ 15: 177–187 Article CAS PubMed Google Scholar * Aydin A, Genc K, Akhisaroglu M, Yorukoglu K, Gokmen N, Gonullu E 2003 Erythropoietin
exerts neuroprotective effect in neonatal rat model of hypoxic-ischemic brain injury. _Brain Dev_ 25: 494–498 Article PubMed Google Scholar * Kumral A, Baskin H, Gokmen N, Yilmaz O, Genc
K, Genc S, Tatli MM, Duman N, Ozer E, Ozkan H 2004 Selective inhibition of nitric oxide in hypoxic-ischemic brain model in newborn rats: is it an explanation for the protective role of
erythropoietin?. _Biol Neonate_ 85: 51–54 Article CAS PubMed Google Scholar * Levine S 1960 Anoxic-ischemic encephalopathy in rats. _Am J Pathol_ 36: 1–17 CAS PubMed PubMed Central
Google Scholar * Rice JE 3rd, Vannucci RC, Brierley JB 1981 The influence of immaturity on hypoxic-ischemic brain damage in the rat. _Ann Neurol_ 9: 131–141 Article PubMed Google Scholar
* Koelfen W, Freund M, Varnholt V 1995 Neonatal stroke involving the middle cerebral artery in term infants: clinical presentation, EEG and imaging studies, and outcome. _Dev Med Child
Neurol_ 37: 204–212 Article CAS PubMed Google Scholar * Lynch JK, Hirtz DG, DeVeber G, Nelson KB 2002 Report of the National Institute of Neurological Disorders and Stroke workshop on
perinatal and childhood stroke. _Pediatrics_ 109: 116–123 Article PubMed Google Scholar * Mader I, Schoning M, Klose U, Kuker W 2002 Neonatal cerebral infarction diagnosed by
diffusion-weighted MRI: pseudonormalization occurs early. _Stroke_ 33: 1142–1145 Article PubMed Google Scholar * Juul SE, Stallings SA, Christensen RD 1999 Erythropoietin in the
cerebrospinal fluid of neonates who sustained CNS injury. _Pediatr Res_ 46: 543–547 Article CAS PubMed Google Scholar * Wen TC, Rogido M, Gressens P, Sola A 2004 A reproducible
experimental model of focal cerebral ischemia in the neonatal rat. _Brain Res Brain Res Protoc_ 13: 76–83 Article PubMed Google Scholar * Wen TC, Rogido M, Genetta T, Sola A 2004
Permanent focal cerebral ischemia activates erythropoietin receptor in neonatal rat brain. _Neurosci Lett_ 355: 165–168 Article CAS PubMed Google Scholar * Cattaneo E, Conti L, De-Frajac
1999 Signalling through the JAK-STAT pathway in the developing brain. _Trends Neurosci_ 22: 365–369 Article CAS PubMed Google Scholar * Socolovsky M, Fallon AE, Wang S, Brugnara C,
Lodish HF 1999 Fetal anemia and apoptosis of red cell progenitors in Stat5a−/−5b−/− mice: a direct role for Stat5 in Bcl-X(L) induction. _Cell_ 98: 181–191 Article CAS PubMed Google
Scholar * Digicaylioglu M, Lipton SA 2001 Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kB signalling cascades. _Nature_ 412: 641–647 Article CAS PubMed
Google Scholar * Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS Jr 1998 NF-kB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation.
_Science_ 281: 1680–1683 Article CAS PubMed Google Scholar * Pettmann B, Henderson CE 1998 Neuronal cell death. _Neuron_ 20: 633–647 Article CAS PubMed Google Scholar * Sun Y, Zhou
C, Polk P, Nanda A, Zhang JH 2004 Mechanisms of erythropoietin-induced brain protection in neonatal hypoxia-ischemia rat model. _J Cereb Blood Flow Metab_ 24: 259–270 Article CAS PubMed
Google Scholar * Wen TC, Tanaka J, Peng H, Desaki J, Matsuda S, Maeda N, Fujita H, Sato K, Sakanaka M 1998 Interleukin 3 prevents delayed neuronal death in the hippocampal CA1 field. _J Exp
Med_ 188: 635–649 Article CAS PubMed PubMed Central Google Scholar * Laemmli UK 1970 Cleavage of structural proteins during the assembly of the head of bacteriophage T4. _Nature_ 227:
680–685 Article CAS PubMed Google Scholar * Towbin H, Staehelin T, Gordon J 1979 Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and
some applications. _Proc Natl Acad Sci USA_ 76: 4350–4354 Article CAS PubMed PubMed Central Google Scholar * Kira M, Tanaka J, Sobue K 1995 Caldesmon and low Mr isoform of tropomyosin
are localized in neuronal growth cones. _J Neurosci Res_ 40: 294–305 Article CAS PubMed Google Scholar * Tanaka J, Fujita H, Matsuda S, Toku K, Sakanaka M, Maeda N 1997 Glucocorticoid-
and mineralocorticoid receptors in microglial cells: the two receptors mediate differential effects of corticosteroids. _Glia_ 20: 23–37 Article CAS PubMed Google Scholar * Rademakers
RP, van der Knaap MS, Verbeeten B Jr, Barth PG, Valk J 1995 Central cortico-subcortical involvement: a distinct pattern of brain damage caused by perinatal and postnatal asphyxia in term
infants. _J Comput Assist Tomogr_ 19: 256–263 Article CAS PubMed Google Scholar * Volpe JJ 2001 Perinatal brain injury: from pathogenesis to neuroprotection. _Ment Retard Dev Disabil Res
Rev_ 7: 56–64 Article CAS PubMed Google Scholar * Joshi CN, Jain SK, Murthy PS 2004 An optimized triphenyltetrazolium chloride method for identification of cerebral infarcts. _Brain Res
Brain Res Protoc_ 13: 11–17 Article CAS PubMed Google Scholar * Lipton P 1999 Ischemic cell death in brain neurons. _Physiol Rev_ 794: 1431–1568 Article Google Scholar * Kumral A,
Uysal N, Tugyan K, Sonmez A, Yilmaz O, Gokmen N, Kiray M, Genc S, Duman N, Koroglu TF, Ozkan H, Genc K 2004 Erythropoietin improves long-term spatial memory deficits and brain injury
following neonatal hypoxia-ischemia in rats. _Behav Brain Res_ 153: 77–86 Article CAS PubMed Google Scholar * Dittmar M, Spruss T, Schuierer G, Horn M 2003 External carotid artery
territory ischemia impairs outcome in the endovascular filament model of middle cerebral artery occlusion in rats. _Stroke_ 34: 2252–2257 Article PubMed Google Scholar * Junk AK, Mammis
A, Savitz SI, Singh M, Roth S, Malhotra S, Rosenbaum PS, Cerami A, Brines M, Rosenbaum DM 2002 Erythropoietin administration protects retinal neurons from acute ischemia-reperfusion injury.
_Proc Natl Acad Sci USA_ 99: 10659–10664 Article CAS PubMed PubMed Central Google Scholar * Shingo T, Sorokan ST, Shimazaki T, Weiss S 2001 Erythropoietin regulates the in vitro and in
vivo production of neuronal progenitors by mammalian forebrain neural stem cells. _J Neurosci_ 21: 9733–9743 Article CAS PubMed PubMed Central Google Scholar * Leist M, Ghezzi P, Grasso
G, Bianchi R, Villa P, Fratelli M, Savino C, Bianchi M, Nielsen J, Gerwien J, Kallunki P, Larsen AK, Helboe L, Christensen S, Pedersen LO, Nielsen M, Torup L, Sager T, Sfacteria A,
Erbayraktar S, Erbayraktar Z, Gokmen N, Yilmaz O, Cerami-Hand C, Xie QW, Coleman T, Cerami A, Brines M 2004 Derivatives of erythropoietin that are tissue protective but not erythropoietic.
_Science_ 305: 239–242 Article CAS PubMed Google Scholar * Kawakami M, Sekiguchi M, Sato K, Kozaki S, Takahashi M 2001 Erythropoietin receptor-mediated inhibition of exocytotic glutamate
release confers neuroprotection during chemical ischemia. _J Biol Chem_ 276: 39469–39475 Article CAS PubMed Google Scholar * Marti HH 2004 Erythropoietin and the hypoxic brain. _J Exp
Biol_ 207: 3233–3242 Article CAS PubMed Google Scholar * Campana WM, Myers RR 2003 Exogenous erythropoietin protects against dorsal root ganglion apoptosis and pain following peripheral
nerve injury. _Eur J Neurosci_ 18: 1497–1506 Article PubMed Google Scholar Download references AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Division of Neonatal-Perinatal Medicine,
Department of Pediatrics, Emory University School of Medicine, Atlanta, 30322, Georgia Augusto Sola, Marta Rogido, Ben H Lee, Tom Genetta & Tong-Chun Wen Authors * Augusto Sola View
author publications You can also search for this author inPubMed Google Scholar * Marta Rogido View author publications You can also search for this author inPubMed Google Scholar * Ben H
Lee View author publications You can also search for this author inPubMed Google Scholar * Tom Genetta View author publications You can also search for this author inPubMed Google Scholar *
Tong-Chun Wen View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Tong-Chun Wen. ADDITIONAL INFORMATION This study was
supported in part by grants for scientific research from the Children's Research Center (T.C.W.) and the Goddard Scholarship (A.S.), Emory University (Atlanta, GA). RIGHTS AND
PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Sola, A., Rogido, M., Lee, B. _et al._ Erythropoietin after Focal Cerebral Ischemia Activates the Janus
Kinase–Signal Transducer and Activator of Transcription Signaling Pathway and Improves Brain Injury in Postnatal Day 7 Rats. _Pediatr Res_ 57, 481–487 (2005).
https://doi.org/10.1203/01.PDR.0000155760.88664.06 Download citation * Received: 23 June 2004 * Accepted: 03 September 2004 * Issue Date: 01 April 2005 * DOI:
https://doi.org/10.1203/01.PDR.0000155760.88664.06 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link
is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative