Ar coactivators, cbp/p300, are critical mediators of dna repair in prostate cancer
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Castration resistant prostate cancer (CRPC) remains an incurable disease stage with ineffective treatments options. Here, the androgen receptor (AR) coactivators CBP/p300, which are
histone acetyltransferases, were identified as critical mediators of DNA damage repair (DDR) to potentially enhance therapeutic targeting of CRPC. Key findings demonstrate that CBP/p300
expression increases with disease progression and selects for poor prognosis in metastatic disease. CBP/p300 bromodomain inhibition enhances response to standard of care therapeutics.
Functional studies, CBP/p300 cistrome mapping, and transcriptome in CRPC revealed that CBP/p300 regulates DDR. Further mechanistic investigation showed that CBP/p300 attenuation via
therapeutic targeting and genomic knockdown decreases homologous recombination (HR) factors in vitro, in vivo, and in human prostate cancer (PCa) tumors ex vivo. Similarly, CBP/p300
expression in human prostate tissue correlates with HR factors. Lastly, targeting CBP/p300 impacts HR-mediate repair and patient outcome. Collectively, these studies identify CBP/p300 as
drivers of PCa tumorigenesis and lay the groundwork to optimize therapeutic strategies for advanced PCa via CBP/p300 inhibition, potentially in combination with AR-directed and DDR
therapies. SIMILAR CONTENT BEING VIEWED BY OTHERS ESS2 CONTROLS PROSTATE CANCER PROGRESSION THROUGH RECRUITMENT OF CHROMODOMAIN HELICASE DNA BINDING PROTEIN 1 Article Open access 31 July
2023 A NONCANONICAL AR ADDICTION DRIVES ENZALUTAMIDE RESISTANCE IN PROSTATE CANCER Article Open access 09 March 2021 TARGETING CDCP1 GENE TRANSCRIPTION COACTIVATED BY BRD4 AND CBP/P300 IN
CASTRATION-RESISTANT PROSTATE CANCER Article 05 May 2022 INTRODUCTION Prostate cancer (PCa) remains the second most common cause of male cancer mortality in the USA [1, 2]. The androgen
receptor (AR), a hormone-activated transcription factor, plays vital roles in the development and progression of PCa. Thus, androgen-deprivation therapy (ADT) is a standard-of-care
first-line therapy for metastatic PCa. Resistance to ADT leads to almost uniformly lethal disease, termed castration-resistant prostate cancer (CRPC) [3,4,5,6,7]. Since the introduction of
hormonal therapy ~60 years ago [8], progress in developing definitive treatments for aggressive disease stage has been difficult despite major advances in understanding prostate
carcinogenesis and disease biology [9, 10]. Recent development of abiraterone, enzalutamide, apalutamide, darolutamide, radium-223, PSMA-Lu, and cabazitaxel have improved outcome, but
metastatic PCa remains a uniformly fatal disease [11,12,13,14]. Moreover, while molecular subtyping affords therapeutic benefit and improved patient survival in other tumor types, some work
has been done in PCa including PAM50 subtyping and presence DNA repair alterations, but additional work remains to be achieved for more detailed subtyping in PCa [15,16,17,18,19]. The
majority of patients with metastatic PCa are treated identically, without selection of appropriate therapeutic regimens based on tumor profile, and there is no durable therapy for metastatic
disease. Whereas local disease can be effectively treated through radical prostatectomy or radiotherapy [3, 20], non-organ confined disease presents a significant clinical challenge.
First-line treatment for disseminated disease consists of androgen deprivation therapy (ADT), as PCa cells are exquisitely dependent on androgen receptor (AR) signaling for growth and
survival [3, 12, 13, 21]. This regimen is often complemented with the use of direct AR antagonists [9, 10, 22,23,24,25,26]. ADT is initially effective in most patients, and successful
ablation of AR activity is validated by loss of detectable prostate specific antigen (PSA). Notably, PSA is a well-defined AR target gene, and since the protein product is secreted into the
sera, it is a convenient biochemical readout of prostate-specific AR function. ADT results in a heterogeneous cellular response of tumor cell quiescence and cell death, resulting in cancer
remission [9, 12, 23, 24, 27, 28]. Unfortunately, this effect is transient and lasts only 2–3 years, at which time the incurable form of the disease, castration resistant prostate cancer
(CRPC), emerges [3, 13, 14]. It is well established that this transition is largely driven by inappropriate AR reactivation despite the continuation of ADT, leading to patient morbidity [3,
13, 28,29,30,31]. Thus, it is critical to define alternative, complementary strategies that can act in concert with AR-directed therapeutics to suppress CRPC growth and progression. CBP/p300
are paralogous, highly conserved histone acetyltransferases (HAT) that serve as transcriptional co-activators [32,33,34]. Each harbors domains that interact with sequence-specific
transcription factors (including AR and c-Myc). Notably, high p300 expression has been associated with locally advanced disease and castration-resistant AR function [32,33,34]. Previous
studies have demonstrated that CBP expression is also elevated in clinical specimens as a function of disease progression [35]. Furthermore, studies using reporter assays nominated CBP/p300
as coregulators in support of AR and c-Myc [32, 33, 36,37,38,39,40]. In PCa models, CBP/p300 expression is induced in response to androgen ablation, suggesting that CBP/p300 may support
disease progression by amplifying basal AR activity in the castration setting, and thereby enhance tumor progression. Given the potential of CBP/p300 as therapeutic targets, especially for
malignancies that are driven by CBP/p300-dependent transcription factors, CBP/p300 functional activities have been nominated as a possible node of intervention. Previous attempts focused on
suppression of the HAT activity of CBP/p300 [34, 40,41,42,43,44,45,46,47,48,49,50], but these strategies proved ineffective in the preclinical setting. Here, this collaborative study shows
that CBP/p300 expression is enhanced in advanced disease and associated with poor outcome. Furthermore, CBP/p300 correlate closely with AR gene expression and AR activity score in primary
PCa and CRPC. By employing clinically relevant PCa models, the clinical significance of CBP/p300 expression in PCa patients as well as mechanistic evaluation of CBP/P300 transcriptional
reprogramming and DNA damage response pathways were investigated. Findings revealed that CBP/p300 bromodomain suppression sensitizes to AR-dependent DNA-repair. Transcriptional mapping
identified CBP/p300 as regulators of cell proliferation and DNA repair processes, which were functionally confirmed across PCa model systems. To assess relevance, exogenous challenge with
genotoxic stress (utilizing in vitro systems, in vivo models, and human tumors ex vivo) revealed that the CBP/p300 bromodomain is required for AR-mediated DNA repair, and CBP/p300 expression
is linked to DNA repair capacity in the clinical setting. Molecular analyses revealed that CBP/p300 facilitate double-strand break (DSB) repair efficiency via homologous recombination (HR)
mediated DNA damage response (DDR). Congruently, CBP/p300 strongly correlated with HR gene expression in PCa patient tissue. These collective findings reveal that CBP/p300 govern rapid
repair of DNA DSBs by regulating HR gene expression, thus modulating genome integrity, and promoting CRPC growth. In sum, these studies identify CBP/p300 as a driver of PCa tumorigenesis
through coordinated control of critical transcriptional events and lay the groundwork to optimize therapeutic strategies for advanced PCa via CBP/p300 inhibition, potentially in combination
with AR-directed therapies and DDR agents to enhance patient outcome. RESULTS CBP/P300 EXPRESSION IS ENHANCED IN ADVANCED DISEASE AND SELECTS FOR WORSE OUTCOME IN METASTATIC DISEASE Several
PCa studies have highlighted the importance of AR-mediated DNA repair factor regulation, yet this critical facet of AR signaling is incompletely defined. In response to androgen stimulation,
AR regulates a vast transcriptional network as illustrated by cistrome and transcriptome mapping. Furthermore, AR-dependent DNA repair factor regulation is a major effector of the response
to DNA damage [51,52,53,54]. Thus, the AR signaling axis is a key component to further target for potential mechanistic intervention to enhance patient outcome. Importantly, CBP/p300 are
paralogous, highly conserved histone acetyltransferases that serve as transcriptional co-activators for AR [55, 56]. To determine the expression level and importance of CBP/p300 and AR in
disease progression, hormone sensitive prostate cancer (HSPC) and castration resistant prostate cancer (CRPC) patient tissue from publicly available datasets on cBioPortal (TCGA, MSK/DFCI,
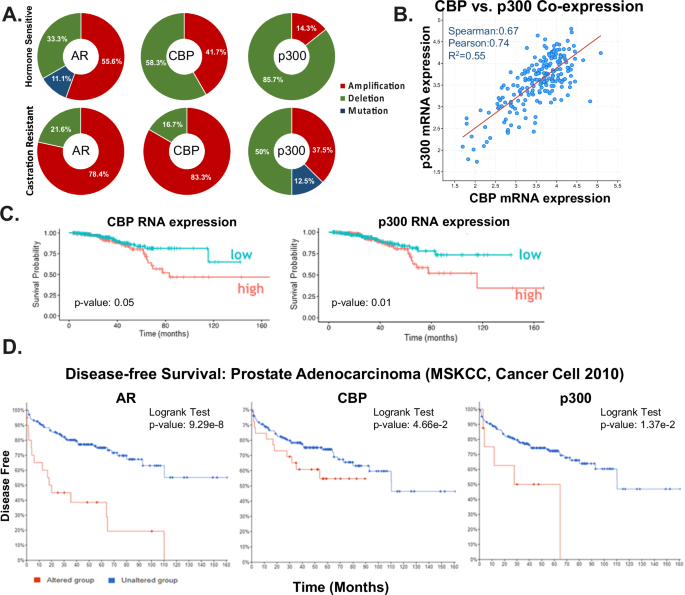
(Nature Genetics 2018), and SU2C/PCF Dream Team (PNAS 2015, 2019)) were examined. In the HSPC cohort, the total alterations of patients harboring AR, CBP, and p300 alterations included 9
patients with AR alterations, 12 patients with CBP alterations, and 7 patients with p300 alterations. Additionally, in the CRPC cohort, the total alterations of patients harboring AR, CBP,
and p300 alterations included 252 patients with AR alterations, 36 patients with CBP alterations, and 8 patients with p300 alterations. _AR_ is amplified in both HSPC (55.6%) and CRPC
(78.4%) (Fig. 1A, Supplementary Fig. 1A). _CBP_ is characterized by 58.3% mutation and 41.7% amplification in HSPC. While _p300_ is characterized by 85.7% mutation, and 14.3% amplification
in HSPC (Fig. 1A). Importantly, with disease progression, patients with CRPC tumors harboring AR, CBP, and p300 alterations are characterized by frequent amplification of CBP and p300 at
83.3% and 37.5%, respectively. Similar increases in amplification of CBP/p300 are observed in additional data sets from MSK/DFCI, Nature Genetics 2018 and SU2C/PCF Dream Team Cell 2015 on
cBioPortal (Supplementary Fig. 1B). Furthermore, CBP and p300 mRNA expression are significantly correlated (spearman = 0.67) in metastatic disease in the SU2C/PCF Dream Team cBioPortal
cohort (Fig. 1B, Supplementary Fig. 1C) indicating that the expression of these two key AR modulators is linked in advanced disease. Additionally, CBP expression is also elevated in clinical
specimens as a function of disease progression as shown in our previous study [35]. The study showed that the immunohistochemistry (IHC) analysis of CBP in HSPC needle biopsy displays lower
levels of CBP compared to CRPC bone marrow transplant which exhibits a significant (p < 0.001) increase in CBP expression. Subsequently, heightened levels of CBP/p300 expression are
associated with unfavorable patient outcomes. Notably, survival probability significantly decreases with higher expression of CBP (p = 0.05) and p300 (p = 0.01) (Fig. 1C). This observed
trend is further supported by cBioPortal data from the Prostate Adenocarcinoma (MSKCC, Cancer Cell, 2010) dataset (Fig. 1D, Supplementary Fig. 1D). Analyzing the correlation between altered
AR, CBP, and p300 and disease-free survival, patients with altered AR (n = 20), CBP (n = 29), and p300 (n = 8) exhibit significantly decreased survival probability, indicated by p-values of
9.29−8, 4.66e−2, and 1.37e−2, respectively. In terms of disease-free survival, patients with alterations in AR, CBP, and p300 experienced a maximum of 110, 90, and 65 months, while some
patients without these alterations surpassed >160 months of disease-free survival (Fig. 1D). Importantly, the correlation of CBP/p300 with disease progression and poor patient outcome
were consistently replicated in various datasets, (Supplementary Fig. 1D). Combined, these studies implicate amplification and thus functional induction of CBP/p300 as an effector of disease
progression. CBP/P300 BROMODOMAIN SUPPRESSION ENHANCES RESPONSE TO STANDARD OF CARE THERAPEUTICS Given the potential of CBP/p300 as therapeutic targets, especially for malignancies driven
by CBP/p300-dependent transcription factors, CBP/p300 functional activities have been proposed as a conceivable target of intervention. Previous attempts focused on suppression of the HAT
activity of CBP/p300 [34, 40,41,42,43,44,45,46,47,48,49,50], but these strategies proved ineffective in the preclinical setting. Our initial study [35] utilizing CCS1477 (Inobrodib) that
selectively targets the CBP/p300 bromodomain, which recognizes acetylated lysine residues, demonstrated CBP/p300 bromodomain inhibition impacts AR and c-Myc signaling, resulting in decreased
tumor growth in CRPC [35, 36, 42, 45]. As previously reported, CCS1477 binds to the bromodomain of CBP/p300 and inhibits tumor growth by disrupting AR signaling in vitro, in vivo, and ex
vivo [35]. In the present study, we aimed to further elucidate the mechanism(s) of action of CBP/p300 in PCa and explore the potential clinical application of CCS1477. First, we validated
the effect of CBP/p300 bromodomain inhibition by exposing CRPC cell model systems to 1 mM CCS1477 over time. CRPC cells exposed to CCS1477 were harvested at 0, 4, 24, and 48 h to determine
the impact on the AR signaling axis via AR, AR-SV (splice variants—AR-V7), c-MYC, CBP, and p300 expression. Consistent with our previous study, AR isoforms, and c-MYC protein expression
decreased over time while CBP and p300 expression remained stable (Fig. 2A). This validated previous findings that CBP/p300 inhibition decreased AR signaling suggesting altered function of
CBP/p300 without impacting protein expression. The biological effect of CBP/p300 on cell cycle progression in CRPC was assessed in multiple cell models, wherein CBP/p300 bromodomain
inhibition resulted in decreased S phase and G2/M arrest over time (Fig. 2B, Supplementary Fig. 2A). As an important cellular checkpoint, G2/M arrest is associated with DNA damage repair
(DDR). In accordance, key DDR and cell cycle markers including _c-MYC_, _TP53_, and _CDKN1A_ mRNA expression decreased with CCS1477 at 0, 12, 24, and 48 h in CRPC models (Supplementary Fig.
2B). Thus, tumor-associated AR coactivators, CBP/p300, appear to be essential for cellular proliferation. To examine the biological relevance, combinatorial studies with standard-of-care
(SOC) therapies and CBP/p300 inhibition were performed for downstream cell survival assessment. Specifically, SOC therapies such as irradiation (IR), cisplatin (platinum-based therapy),
olaparib (PARP inhibitor), and doxorubicin were assessed in combination with increasing concentrations of CCS1477. Importantly, combination therapy at the highest doses were the most lethal
in CRPC models (Fig. 2C–E, Supplementary Fig. 2C–E). Specifically, the lowest relative cell growth is observed in CRPC cells treated with CCS1477 and higher doses of SOC with enhanced
combination index achieved in these combinatorial studies indicating synergy (Fig. 2C–D and Supplementary Fig. 2C–D). The IC50 values decreased 5–10-fold from single agent (CCS1477) to
combination with SOC in 22Rv1 and C4-2 cells (Fig. 2D). To understand the effect of the combination therapy on the AR signaling axis, AR, c-MYC, CBP, and p300 protein expression was examined
in CRPC cell lines treated with 5 Gy IR, 1 mM cisplatin, 2.5 mM olaparib, and 0.5 mM doxorubicin alone and in combination with CCS1477 (Fig. 2E, Supplementary Fig. 2E). Consistently, in
both cell lines, AR and c-Myc expression decreased further in combination treatment than with single agent. Interestingly, CBP/p300 expression varies between the different SOC therapies
alone compared to combination. Notably, p300 protein expression decreases with CCS1477 + 5 Gy IR in 22Rv1 and C4-2 cells compared to 5 Gy IR alone. In sum, CCS1477 demonstrates promising
potential as a novel small molecule inhibitor of CBP/p300 through a synergistic effect in combination with existing therapies for CRPC. CBP/P300 BROMODOMAIN INHIBITION REDUCES THE AR AND
CBP/P300 CISTROMIC LANDSCAPE These remarkable responses strongly underscore the importance of further understanding the tumorigenic role of the CBP/p300 bromodomain in cancer progression.
However, genome-wide understanding of CBP/p300 function is not yet fully defined in PCa. Thus, the mechanism through which CBP/p300 influences aggressive disease was assessed. AR function
was evaluated via chromatin immunoprecipitation followed by DNA sequencing (ChIP-Seq) with CCS1477. CBP/p300 cistrome mapping in CRPC, along with AR, using a stringent cutoff identified
31137, 35775, and 3581 binding sites of AR, CBP, and p300, respectively in 22Rv1 cells. Interestingly, in the presence of CBP/p300 bromodomain inhibition, the cistromic landscape is reduced
to 19300, 11920, and 1660 AR, CBP, and p300-bound sites, respectively (Fig. 3A, Supplementary Fig. 3A–B). This decrease in AR, CBP, and p300 binding suggests a putative loss of function in
AR function when CBP/p300 is inhibited, likely contributing to the biological changes seen in Fig. 2. Decreased AR, CBP, and p300 have previously shown to change promotor binding [32, 57,
58]. As CCS1477 treatment decreased AR function and cistromic landscape, genomic annotation of AR, CBP, and p300 binding sites were performed using cis-regulatory element annotation system
(CEAS) package to elucidate a description of impacted regulatory sites (Fig. 3B). As shown in Fig. 3B, CBP/p300 bromodomain inhibition did not significantly alter AR regions of binding, with
majority of binding observed at intronic (45%) and intergenic (44%) regions, followed by promoters (6%) with or without CBP/p300 inhibition (Fig. 3B—top). Interestingly, CCS1477 treatment
shifted the regions of binding for CBP from predominately intergenic (42%), intronic (28%), and promoter (24%) to predominantly promoter (34%), intergenic (31%), and then intronic (29%)
regions (Fig. 3B—middle). Conversely, inhibition of CBP/p300 bromodomain enhanced p300 binding towards intergenic (from 36% to 45% with CCS1477) and intronic (from 33% to 37% with CCS1477)
regions. Additionally, CCS1477 treatment results in decreased binding to the promoter (26–16% with CCS1477) regions (Fig. 3B—bottom), suggesting that p300 binding shifts towards intergenic
and intronic regions in CRPC following CBP/p300 inhibition. This suggests that CBP and p300 might have distinct regulatory activity which were next assessed with motif analyses to determine
potential binding partners. Specifically, to discern potential transcription factors associated with AR, CBP, and p300, known-motif analysis was conducted using a broad window around the
center of binding (1 KB) (Fig. 3C, Supplementary Fig. 3C). Expected enrichment of AR binding elements (ARE, GRE) and key pioneer factors (FOXA1, FOXA2, FOXM1) was complemented by enrichment
of cancer-associated transcription factors of PCa relevance, including several with oncogenic activity (e.g., NF1, HOXB13, ETS factors) [59,60,61,62,63,64]. Specifically, enriched motifs
correspond to components of the FoxA1/AR complex or are directly driven by AR to promote AR signaling, cell proliferation/invasion, metabolic rewiring, and ultimately tumor growth (Fig.
3C—right). Analysis of known CBP binding motifs showed prevalent binding to FOXA1, FOXM1, FOXO1, ELK1, and NF1 (Fig. 3C—middle). Similarly, analyses of known p300 binding motifs depicted
binding to the FOXM1, FOXA1, FOXO3, FOXA3A, and THAP (Fig. 3C—left). To further investigate the potential mechanisms of CBP/p300 function, de novo motif analysis was assessed using a window
of 50 bp adjacent to the center of binding (Fig. 3D). In addition to motifs for Forkhead Box proteins (e.g., FOXM1), multiple motifs of PCa relevance were enriched proximal to AR binding,
including several factors elevated in PCa, linked with androgen-associated cancer growth, interleukin regulation, and cancer progression (e.g., NFIA, GRE, and Arid5a) [65, 66] (Fig. 3D—top).
Importantly, additional motifs were enriched proximal to CBP and p300 binding, including another Forkhead Box protein (FOXA1), a key pioneer factor pivotal to PCa progression (Fig. 3D) [60,
63]. Further, CBP was enriched for PCa-associated transcription factors, including ETS-factor, Elk4, and SP1 [62, 64, 67,68,69,70]. Conversely, in addition to FOXA1, p300 was enriched for
mitochondrial factor (GFY), zinc finger proteins (Bc111a, ZNF143), and PCa-associated transcription factors (HOXB13) [71,72,73,74,75,76,77,78]. Pathway analysis utilizing Hallmarks and KEGG
gene set enrichment analyses (GSEA) identified that CBP and p300-bound sites impact key hallmarks of cancers including cell cycle, DNA repair, and metabolic signaling (Fig. 3E, Supplementary
Fig. 3D–E). Transcriptional signatures also identified in this study highlight pathways of functional importance. In sum, these observed enrichments and related pathway analyses (Fig. 3E,
Supplementary Fig. 3D–E) provide the first insight into genome-wide CBP/p300 activity in CRPC models and reveal potential exclusive oncogenic functions of the coactivator partners beyond AR
signaling axis. These data provide insights into the potential mechanism driving AR signaling and CBP/p300 cofactor alterations in CRPC. CBP/P300 GOVERN TRANSCRIPTIONAL NETWORKS CRITICAL FOR
RESPONSE TO DNA DAMAGE The biological significance of the AR signaling axis in driving progression to CRPC is well established [6, 12, 21, 22, 28, 30, 35, 40, 41, 43, 45, 48, 79], and thus
AR remains the most preferred target in treating PCa. However, with the observation that AR co-activators CBP/p300 are associated with poor outcomes in metastatic PCa [35] (Fig. 1), coupled
with linkage of the newly identified CBP/p300 cistrome to cancer-promoting pathways (Fig. 3), it is now cogent that CBP/p300 play pivotal and potentially targetable roles in PCa. Previous
studies identified that CBP/p300 inhibition via CCS1477 resulted in significant transcriptional alterations including androgen response and MYC target pathways [35] (Supplementary Fig. 4A).
In this study, we performed genome-wide assessment of the CBP- and p300-sensitive transcriptional networks utilizing newly generated doxycycline-regulated isogenic paired model systems of
inducible _CBP_ and _p300_ knockdown. As shown, ~80% ablation of CBP and p300 protein was achieved after shRNA induction (Fig. 4A—top), subsequent to which the whole-transcriptome analysis
of CBP and p300 individually was assessed in human CRPC models (Fig. 4A—bottom). Principal component analyses (PCA) indicated a high level of concordance between biological replicates as
shown in sample clustering within treatment groups with strong consistency amongst biological replicates (Supplementary Fig. 4B). Major transcriptional changes were observed (1831
upregulated, 2785 downregulated genes) after CBP knockdown (adjusted _p_-value < 0.001, fold change > 2) and (3219 upregulated, 3459 downregulated genes) after p300 knockdown (adjusted
_p_-value < 0.001, fold change > 2) indicating that CBP and p300 influence large gene networks. Gene set enrichment analysis (GSEA) (Fig. 4B, Supplementary Fig. 4C) revealed that
CBP/p300 govern transcriptional programs of oncogenic relevance, including those involved in DNA replication, cell cycle regulation, and multiple DNA repair processes. The biological effect
of CBP and p300 on cell cycle progression in CRPC was assessed in multiple distinct isogenic pairs, wherein CBP and p300 individual ablation resulted in G2/M arrest (Fig. 4C, Supplementary
Fig. 4D) in the presence of genotoxic insult, i.e., irradiation (IR). These studies reveal a requirement of CBP/p300 for cell cycle progression in CRPC. Thus, tumor-associated CBP/p300
appear to be essential for cellular proliferation thus further highlighting a therapeutic window. Downstream analyses demonstrated the biological relevance, as evidenced by a 1.6–1.8-fold
reduction in surviving cells after exposure to 5 Gy IR in CBP-depleted and p300-depleted cells (Fig. 4E) and decreased cell growth after DNA damage from SOC (cisplatin, olaparib (PARPi), and
doxorubicin) treated cells with CBP/p300 ablation (Fig. 4F–G). Specifically, the lowest relative cell growth is observed in CRPC cells treated with single agent SOC in combination with CBP
and p300 knockdown (Fig. 4E–G). The IC50 values decreased 2-5-fold from single agent (SOC) to combination with CBP-depletion or p300-depletion in 22Rv1 and C4-2 cells (Fig. 4G). These
findings reveal new insight into CBP/p300 function as a modulator of cell cycle checkpoint control and cell proliferation in response to DSB and implicate CBP/p300 in promoting cancer cell
survival. CBP/P300 MODULATES DNA REPAIR FACTOR EXPRESSION AND HOMOLOGOUS RECOMBINATION Since both CBP and p300 expression is elevated with disease progression and associated with worse
clinical outcomes (Fig. 1), combined with molecular observations that CBP/p300 perturbations result in transcriptional rewiring influencing DNA repair (Figs. 3–4), unbiased computational
strategies were employed to discern the potential mechanism of action by which CBP/p300 influence DNA damage response. To prioritize the genes for downstream analyses, genes significantly
altered with stringent cutoffs (p < 0.01, fold change >2.0) were compared between CBP and p300 ablation (i.e., shCBP and shP300 conditions) (Fig. 5A). This overlay identified 3000
genes in common with both CBP and p300 knockdowns in 22Rv1 cells. Next, these 3000 genes were compared to DNA damage repair (DDR) genes using MSigDB, which have been previously curated and
published [80]. This resulted in a list of CBP/p300-regulated DDR genes, which were further grouped into the main DDR pathways including HR (homologous recombination), BER (base excision
repair), MMR (mismatch repair), NER (nucleotide excision repair), and NHEJ (non-homologous end joining) to determine which DDR pathway is most significantly regulated by CBP/p300. These
analyses revealed potentially critical roles for CBP/p300 in regulating factors associated with HR (23% of CBP/p300-regulated genes), BER (19%), MMR (15%), NER (14%), and NHEJ (1%).
Concurrently, transcriptional profiling following treatment with CCS1477 revealed that CBP/p300 regulates HR factors (Supplementary Fig. 5B). CBP/p300 ablation and inhibition significantly
impacted HR-mediated repair as functional CBP/p300 signaling is required for expression of major HR factors, including _BRCA2, POLD2, RAD51_, and _XRCC2_ (Fig. 5B, Supplementary Fig. 5B).
These findings suggest that CBP/p300 modulate factors associated with HR-mediate DNA damage, impacting disease aggressiveness and response to standard-of-care. CBP/P300 ATTENUATION DECREASES
HR EFFICIENCY An HR reporter assay was conducted by utilizing a well-established U2OS cell line to read HR repair efficiency through revived GFP expression [81, 82] (Fig. 6A—Top). CBP/p300
inhibition with 2.5 µM CCS1477 decreased HR repair by (8-fold) when compared to ATM inhibition (6-fold change) (Fig. 6A—Middle). Similar results were observed with individual and combined
knockdown of CBP and p300 as compared to ATM inhibition. Overall, all inhibited cells had significantly (p < 0.001) decreased HR efficiency than control cells (Fig. 6A—Bottom). These
findings nominate CBP/p300 as positive effectors of HR. To assess the impact of these findings on PCa, CBP/p300 isogenic pairs with individual knockdown of CBP and p300 in the presence of
genotoxic insult, IR, were assessed for impact on HR target gene expression. HR factors including, _ATM_, _CHEK2_, and _RAD50_, mRNA expression was significantly decreased in knockdown cells
with genotoxic insult (Fig. 6B). To further validate these findings, 22Rv1 and C4-2 cells were treated with CCS1477 and genotoxic insult. HR factors, including _ATM_, _CHEK2_, and _RAD50_
mRNA expression decreased (p < 0.001) compared to IR conditions (Fig. 6C—Top). Similar results were observed in C4-2 cells (p < 0.001) (Fig. 6C—Bottom and Supplementary Fig. 6A).
Concordant changes in pATM and pCHK2 protein levels were observed as a result of CBP/p300 inhibition (Fig. 6D, Supplementary Fig. 6B). In sum, these findings show that CCS1477 destabilizes
the HR mediated repair pathway through CBP/p300 attenuation (e.g., bromodomain inhibition and genomic knockdown). TARGETING CBP/P300 IMPACTS HR-MEDIATED REPAIR AND PATIENT OUTCOME CBP/p300
inhibition holds therapeutic potential in combination with first-line therapy drugs (Fig. 2). Therefore, the impact on HR-mediated repair was assessed in patient samples. Analysis of key HR
genes with CBP/p300 attenuation (inhibition or knockdown) in CRPC cell lines were compared, and top altered HR genes were identified for impact in PCa patients (Fig. 7A). Importantly,
CBP/p300 inhibition in both CRPC models (22Rv1 and C4-2) and isogenic pairs with knockdown revealed that CBP/p300 regulate key HR factors. Next, to further examine the translational
relevance, HR genes (_ATM, MRE11A_, and _RAD50_) were compared for co-occurrence with _CBP_ and _p300_ in human data set from cBioportal (SU2C/PCF Dream Team (PNAS 2019) (Fig. 7B).
Correlations for all three HR genes were statistically significant (p < 0.001), highlighting HR pathway as a strong mode of DDR in patients with elevated CBP/p300. Furthermore, xenograft
models of 22Rv1 cells undergoing 28-days treatment with 20 mg/kg CCS1477 displayed a significant decrease in HR target genes (_MRE11A, RAD50_, _CHEK2_, and _BRCA2_) (Fig. 7C, Supplementary
Fig. 7A) supporting in vitro findings. To assess the potential for therapeutic resistance, impact of long-term treatment with CBP/p300 inhibition was evaluated using patient derived
xenografts (PDX—CP50c, previously published [35]) over a period of 8 days and when tumors reached 300% of their original growth (long-term) of CCS1477 (Fig. 7D). Specifically, for the
long-term treatment arm, mice were allowed to reach maximal volume (300% of their original growth) before they were culled. So, mice in this treatment group were all culled at different time
points. Our previously published study [35] depicted the efficacy results until days 31-35 in which it was reported that there was significant decreased growth with CBP/p300 inhibitor
(CCS1477) treatment. Importantly, short-term CCS1477 treatment decreased HR-mediated repair as measured via expression levels of HR genes (_MRE11A_ and _RAD50_). Moreover, CBP/p300
expression co-occurred with additional HR factors (_ATM, RAD51_, and _RAD54L_) (Supplementary Fig. 7B). Intriguingly, long-term CBP/p300 inhibitor treatment caused an increase in HR genes,
displaying potential resistance to CCS1477 over time (Fig. 7D). Finally, to gain a better understanding of CCS1477 treatment on clinical tumor growth, patient derived explants (PDEs) were
utilized [83]. Briefly, in this ex vivo system, tumor and adjacent non-neoplastic samples from primary PCa patients undergoing radical prostatectomy were placed on dental sponges and treated
with increasing concentrations of CCS1477 (1–5 µM) (Fig. 7E, F). Increasing doses of CCS1477 significantly decreased (p < 0.05) tumor growth as measured by Ki67 staining, a marker of
proliferation (Fig. 7E). Furthermore, PDEs were analyzed for expression of HR genes with CBP/p300 inhibition (Fig. 7F). Consistent with the in vitro cell line and in vivo xenograft and PDX
studies, decreased HR gene expression is observed in ex vivo PDEs with CBP/p300 bromodomain inhibition. Importantly, these findings support the interplay of the CBP/p300 axis and HR-mediated
repair. Analysis of patients treated with CCS1477 indicate that CBP and p300 co-occur with HR targets, _ATM_, _RAD50_, _RAD51_, and _RAD54L_, in PCa patients (Supplementary Fig. 7B).
Combined, these findings identify CBP/p300 attenuation as drivers of HR-mediated anti-tumorigenic signaling in PCa and identify a potential novel target to enhance patient outcome (Fig. 7G).
DISCUSSION CRPC remains an incurable disease stage with ineffective treatments options to avoid lethality. In this study, we elucidate the mechanism of CBP/p300 inhibition functioning
through HR, to potentially enhance therapeutic targeting of CRPC. We previously established CBP and p300 attenuation via bromodomain inhibition and isogenic knockdown models regulate AR
signaling in a c-MYC manner in CRPC in vitro and in vivo models [35]. Now, this study further illuminated the mechanism of action of CBP/p300 inhibition which resulted in decreased PCa
growth by directly impacting HR-mediated DNA repair. Key findings demonstrate that: (i) CBP/p300 expression increases as disease progresses to CRPC and is associated with poor patient
outcome; ii) Inhibition of the CBP/p300 bromodomain enhances response to SOC therapies; iii) Cistromic landscape of CBP and p300 is drastically reduced with CBP/p300 inhibition; iv) CBP/p300
governs pathways critical for the response to DNA damage via transcriptional regulation; v) CBP/p300 regulates HR-mediated repair in vitro, in vivo, and ex vivo; vi) Targeting CBP/p300
impacts HR-mediated repair; and vii) CBP/p300 are strongly associated with HR factor expression and poor outcome in human disease. These studies are the first to highlight the molecular
interplay of AR coactivators, CBP/p300, and HR-mediated repair in CRPC with preclinical and clinically relevant models (Fig. 7G). In sum, these studies nominate potential combinatorial
targets for therapeutic intervention to enhance patient outcome. Previous studies using reporter assays nominated CBP/p300 as coregulators in support of AR and c-Myc [32, 33,
36,37,38,39,40], and in PCa models, CBP/p300 expression is induced in response to androgen ablation, suggesting that CBP/p300 may support disease progression by amplifying basal AR activity
in the castration setting, and thereby enhance tumor progression. This concept is further supported by our previously published [35] study and this current study. CBP and p300 are often
treated as interchangeable due to their functional similarities. However, both proteins have nuanced distinctions that enable unique functions [58, 77]. This study delves into elucidating
these potential differences utilizing cistromic and transcriptomics analyses with CBP/p300 pharmacological and genomic attenuation (Figs. 3–4). Notably, this is the first study to identify
the CBP and p300 cistromes in CRPC models (Fig. 3). Importantly, cistromic analyses in CRPC revealed distinct binding regions of CBP and p300 (Fig. 3B). CBP preferentially binds to the
intergenic region (42%) while p300 binds to the intronic (33%) and promoter (26%) regions, which is shifted with CBP/p300 bromodomain inhibition via CCS1477 treatment. Moreover, while the
transcriptional partners with which CBP/p300 interact with are similar as seen with the FOX family of transcription factors (Fig. 3C), the mechanism of action may be different as seen with
the unique de novo motif analyses (Fig. 3D). Proximal to CBP binding, motifs are enriched for Ets-factors Elk4 and Sp1, which are linked to PCa oncogenic signaling [62, 64, 67,68,69,70].
Intriguingly, proximal to p300 binding, motifs are enriched for GFY, ZNF143, and HOXB13, which are linked to mitochondrial signaling and zinc-finger function [71,72,73,74,75,76,77,78]. These
unique binding regions and motif analyses by the CBP/p300 paralogs could impact distinct downstream signaling and biological function as seen with the Hallmark pathway analyses indicating
distinct functions of CBP and p300 (Fig. 3E). Future studies will investigate the potential exclusive, non-redundant functions of CBP and p300 as potential therapeutic targets. CBP/p300 have
been studied for decades for their HAT functions as a potential therapeutic target for treating cancer [49, 56, 84, 85]. This exploration has uncovered aspects of the CBP/p300 and HAT
potential mechanism of action. The HAT domain has been shown to regulate DDR by regulating reactive oxygen species (ROS) production and function [55, 86,87,88]. While this study utilizes
pharmacological targeting of the bromodomain of CBP/p300, we acknowledge that targeting of the HAT catalytic domains or the CH1/TAZ binding domains are alternative options. Notably,
targeting these domains were not pursued since pharmacological inhibition of the HAT domain has been therapeutically unsuccessful thus far [49, 55, 56, 58]. In our previous paper [35], we
demonstrated that bromodomain inhibition caused DNA repair de-enrichment and G2M checkpoint arrest. Similarly, genomic attenuation of CBP/p300 with isogenic pairs of CBP and p300 knockdown
utilizing shRNA in CRPC models, revealed key DDR and cell cycle pathways decreased with CBP/p300 knockdown (Fig. 4). Thus, the continuity of pharmacological inhibition and genomic decrease
suggest a potential mechanism of action that was further explored in this study. Intriguingly, the potential interplay of CBP/p300 and DDR could be a novel combinatorial therapy. Numerous
studies have demonstrated a collaborative relationship between AR and DDR pathways, as extensively discussed herein [20, 35, 51,52,53,54, 80, 89]. However, a recent investigation by Hasterok
et al. [90] challenged the notion of a direct regulatory role for AR in DDR gene expression. This study revealed that while the AR antagonist enzalutamide effectively suppressed canonical
AR target gene expression in PCa models, it had no impact on DDR gene sets. Furthermore, PRO-Seq analyses indicated that AR is not the primary regulator of DDR gene induction in response to
genotoxic insult via IR. It is crucial to note that the IR dose (6 Gy) and treatment duration (30 min) employed in this study for PRO-Seq analysis differed from those used in our work and
others. The rapid, transient DNA repair response observed in the study [90] contrasts with the prolonged downstream effects of AR on DDR, as evident in our models following exposure to
genotoxic agents including 4–24 h post-IR, doxorubicin, or cisplatin (Figs. 2, 4, and 6). Ongoing research is focused on elucidating the temporal dynamics and molecular mechanisms underlying
DDR gene activation, with a particular emphasis on the role of CBP/p300 in modulating these processes. A comprehensive understanding of these factors is essential for optimizing therapeutic
interventions. Transcriptomic pathway analysis, functional molecular studies, and biological validation herein revealed that CBP/p300 govern a discrete network of transcriptional programs
of cancer relevance, including regulation of DNA repair processes, cell cycle control, and metabolic regulation. While there are studies detailing CBP/p300 in influencing DNA repair, cell
cycle control, and metabolic regulation [55, 86,87,88, 91,92,93,94,95], the role of CBP/p300 in transcriptional regulation of double-strand DNA repair regulation in CRPC models was unknown.
Functional assessment herein demonstrated that CBP/p300 regulates HR-mediated repair in vitro, in vivo, and ex vivo. Briefly, CBP/p300 attenuation via CCS1477 or shRNA knockdown revealed in
the presence of genotoxic insult resulting in double-strand break, HR-mediated repair was impaired and PCa growth decreased (Figs. 4–6). Importantly, AR is a key driver of PCa and known
factor in DDR including regulation via PARP and DNAPK [20, 51, 52, 80, 89]. Importantly, the CRPC models (22Rv1, C4-2) utilized in this study both maintain HR proficiency and are not
hypersensitive to PARP inhibition [89, 96,97,98]. The CRPC models do have varying BRCA1/2 status with 22Rv1 cells exhibiting BRCA1 deficiency and a BRCA2 mutation, while C4-2 cells possess
wild-type BRCA1 but reduced BRCA2 protein levels compared to other PCa cell lines [99, 100]. Thus, by utilizing these CRPC models with distinct genetic backgrounds, we aimed to identify
common regulatory mechanisms influenced by CBP/p300 that are independent of BRCA1/2 status. Focus on shared pathways enabled the investigation of potential therapeutic targets applicable to
a broader patient population. Future studies will delve deeper into the interplay between BRCA status, PARP inhibition, and CBP/p300 alterations to optimize treatment strategies. Moreover,
potential mechanism of action for CBP/p300 promoting HR-mediating repair could be through coactivator function with AR or via modulation of multiple oncogenic transcriptional factors. As
described in the cistromic analyses (Fig. 3), binding enrichment was observed for several oncogenic transcription factors of established PCa relevance, including forkhead factors, ETS
factors, and zinc finger components. Future studies will be directed at determining which of these factors regulate the underpinning mechanisms for CBP/p300 and HR-mediated repair. Finally,
this study further highlights the clinical importance of CBP/p300 alterations in disease progression. Findings herein revealed that CBP/p300 are strongly associated with poor outcome in
human disease (Fig. 1). Also, CBP/p300 regulation of key HR factors has translational relevance with co-occurrence of _CBP_ and _p300_ expression with HR targets (_ATM_, _MRE11_, and
_RAD50_) in metastatic PCa patients (Fig. 7). These observations are critical given that homologous recombination deficiency (HRD) impacts PCa progression and therapeutic efficacy
[101,102,103,104,105,106,107]. Briefly, PARP inhibitors (PARPi) have been tested and approved for BRCA deficient tumors [108,109,110,111,112]. Thus, the role of CBP/p300 in influencing the
response to PARP1/2 inhibitors should also be explored. Given that CBP/p300 are coactivators of AR and attenuation of these factors are critical for HR-mediated DNA repair, CBP/p300 status
may provide further therapeutic insight into tumors that may respond to PARPi. Determining mechanisms to directly antagonize CBP/p300 function in the clinical setting may also be an
important next avenue of investigation. Currently, ongoing clinical trials to determine the efficacy and tolerability of CCS1477 (Inobrodib) include evaluation in solid tumors (clinical
trial identifier NCT03568656) and hematological malignancies [113]. Specifically, an open label phase I/IIa study of CCS1477 as monotherapy and in combination with AR targeting agents will
lead to more insight for the role of HRD impacting efficacy. The clinical development of CBP/p300 inhibitors may face challenges as monotherapies in metastatic PCa. Therefore, drug
combinations of utilizing DDR agents with CBP/p300 inhibition might prove to be more effective. Lastly, there are recent developments of the use of PROTAC as CBP/p300 degraders and examining
their effect in solid tumors [114, 115]. Importantly, future studies exploring mechanisms of resistance are crucial to enhance therapeutic efficacy regardless of the pharmacological means
utilized to target CBP/p300 function. Taken together, studies herein reveal new knowledge of the interplay between CBP/p300 and DNA repair in advanced PCa, wherein CBP/p300 expression is
associated with poor outcome. Unbiased sequencing, molecular interrogation, and biological assessment of CBP/p300 function suggest CBP/p300 are key drivers of HR-mediated repair by revealing
a novel mechanism of action in response to genotoxic insult. Pharmacological and genetic attenuation of CBP/p300 indicated that these factors impact HR efficacy and promote CRPC growth in
preclinical and clinical models. Thus, targeting AR signaling via its coactivators in combination with DDR agents offers a new potential therapeutic strategy that merits evaluation in
advanced PCa clinical trials to enhance patient outcome of this lethal disease. METHODS CELL CULTURE AND REAGENTS 22Rv1 and C4-2 cells were purchased from ATCC, authenticated by ATCC, and
tested for mycoplasma upon thawing of cells. Doxycycline-inducible cell line models to knockdown expression of CBP and p300 (shCON, shCBP, and shP300) were developed as previously described
[35]. All C4-2 derived cell lines were cultured and maintained in Improved Minimum Essential Medium (IMEM) (Thermo Fisher Scientific, 10024CV) supplemented with 5% FBS (fetal bovine serum,
heat inactivated), 1% l-glutamine (2 mmol/l), and 1% penicillin-streptomycin (100 units/ml). All 22Rv1 derived cell lines were cultured and maintained in Roswell Park Memorial Institute
(Gibco RPMI) (ATCC, CATALOG # A1049101) supplemented with 10% FBS, 1% l-glutamine, and 1% penicillin-streptomycin. All cells were cultured at 37 °C at 5% CO2. CBP/p300 bromodomain inhibitor
was developed from CellCentric and Deta [35]. Briefly, CCS1477 was synthesized according to processes described in the International Patent Application, publication number WO2018073586.
TRANSCRIPTOME DATA ANALYSIS OF TWO MCRPC COHORTS (SU2C AND RMH—ROYAL MARSDEN HOSPITAL) Patient cohort was utilized from previously published study [35]. All patients had given written
informed consent and were enrolled in institutional protocols approved by the Royal Marsden (London, UK) ethics review committee (reference no. 04/Q0801/60). Human biological samples were
sourced ethically, and their research use was in accordance with the terms of the informed consent provided as detailed previously [35]. A total of 159 mCRPC transcriptomes generated by the
SU2C–PCF Prostate Cancer Dream Team [116] and 95 mCRPC transcriptomes from patients treated at Royal Marsden Hospital and Institute of Cancer Research (RMH/ICR) were analyzed [117] using
TopHat2-Cufflinks pipeline. Gene expression as fragments per kilobase of transcript per million mapped reads (FPKM) was calculated. CHROMATIN IMMUNOPRECIPITATION (CHIP)-SEQUENCING 22Rv1
cells were plated in hormone-proficient media. ChIP was performed as previously described [15]. Briefly, cells were cross-linked with 1% fresh formaldehyde for 10 min at room temperature.
Chromatin was sheared to 200–700 bp using Active Motif Ultrasonicator for 30 cycles (30 s on, 30 s off). CBP and p300 antibodies were obtained from Cell Signaling (CST). ChIP-Seq was
performed by Active Motif according to their guidelines and protocols including antibody testing, library preparation, and sequencing. ChIP-Seq data is deposited in the NCI GEO at GSE275777.
CHIP-SEQUENCING ANALYSIS Analyses was performed by expert bioinformaticians. Briefly, FASTQ files were assessed for quality using FASTQC v0.11.5. Reads were aligned to the human genome
reference version hg19 using bowtie2 v2.3.2 [71] with default parameters. Peak calling was performed using MACS2 v2.1.1 [72] with combined replicates, utilizing a q < 0.05 cutoff. Common
and exclusive binding datasets were generated through comparison of statistically significant binding regions. Any overlap between regions was considered “common” while those not
statistically significant in indicated compared dataset was considered “exclusive”. ChIP-Seq binding heatmaps and profiles were generated using deepTools v2.5.7 [73]. Peak annotation and
motif analysis performed using Homer v4.10.3 [74] using the parameters indicated. RNA-SEQUENCING 22Rv1 shCON, 22Rv1 shCBP, and 22Rv1 shP300 cells were treated with doxycycline for 5 days to
knockdown expression of CBP or P300 in biological quadruplicate. Additionally, 22Rv1 and C4-2 cells were treated with CBP/p300 inhibitor (100 nM CCS1477 for 24 h) as detailed previously
[35]. RNA was extracted and purified using the miRNeasy kit (QIAGEN, 217004) by following manufacturer’s instructions. Novogene performed RNA sequencing following their company polices.
Briefly, TruSeq Stranded Total RNA Library Prep Gold kit was used to construct the RNA-Seq libraries. NextSeq 500 sequencer from Illumina was utilized to sequence samples using single-end 75
bp reads. RNA-SEQUENCING ANALYSIS FASTQ files were aligned using STAR v2.5.2a against the human genome (hg19) [118, 119]. Read counts for each gene were generated using featureCounts
utilizing Ensembl as reference gene annotation set [120]. Differential gene expression data were generated using DESeq2 v1.12.4 [121]. Gene set enrichment analysis (GSEA) was performed using
HALLMARKS and KEGG gene signatures from the Molecular Signature Database [122]. RNA-Seq data is deposited in the NCI GEO at GSE275778. FLOW CYTOMETRY All 22Rv1 and C4-2 parental and derived
cells were plated at equal densities in hormone-proficient media (i.e., FBS condition). Then follow all treatment conditions described, cells were incubated with BrdU (1:1000) for 2 h prior
to harvesting. Cells were fixed and processed as previously described [123]. At least 10,000 events per sample were assessed. Analysis was performed using InCyte software (Guava) for
cell-cycle profile with BrdU incorporation and PI (propidium iodide). GENE EXPRESSION ANALYSIS All 22Rv1 and C4-2 parental and derived cells were plated at equal densities in
hormone-proficient media (i.e., FBS condition). Trizol (Invitrogen) was used to isolate RNA and SuperScript VILO (Invitrogen) was used to generate cDNA following manufacturer’s instructions.
PowerSybr (Fischer Scientific 43-676-59) and the ABI StepOne Real-Time PCR system were utilized in accordance with manufacturer’s specifications to perform quantitative PCR (qPCR) analyses.
The primers used are depicted in Table 1 below. IMMUNOBLOTTING All 22Rv1 and C4-2 parental and derived cells were plated at equal densities in (i.e., FBS condition). Generation of cell
lysates was described previously [52]. 40–50 mg of lysate was resolved by SDS-PAGE, transferred to PVDF (polyvinylidene fluoride) membrane, and analyzed using the following antibodies at 1:
500 dilution—CBP (D9B6), p300 (D2X6N), 1:1000 dilution—ATM (Cell Signaling Technology 2873), phospho-ATM (Ser1981) (CST 5883S), CHK2 (Bethyl A300-619A), phospho-CHK2 (Thr68) (CST 2661T),
MRE11 (CST 8344T), RAD50 (CST 8344T), RAD51 (Abcam ab63801), XRCC3 (Novus NB100-165), and 1: 5000 – Vinculin (Sigma-Aldrich V9264). PROLIFERATION ASSAYS All 22Rv1 and C4-2 parental and
derived cells were plated at equal densities in (i.e., FBS condition). Cells were treated with either IR (irradiation), CBP/p300 inhibitor (CCS1477, CellCentric) [35], or doxycycline, which
was refreshed every 48 h. Cell number was quantified using the Quanti-IT Pico Green dsDNA assay kit (Thermo Fisher) at the indicated times of treatment. XENOGRAFT ANALYSIS All animals were
housed in pathogen-free facilities. All mouse work was approved by the Axis BioServices Animal Welfare and Ethical Review Committee and conducted under license and within the guidelines of
the Home Office Animals (Scientific Procedures) Act 1986. Prostate xenograft tumors were established by subcutaneous injection of 22Rv1 cells into non-castrated male athymic nude mice. Once
tumors had reached approximately 150 mm3, animals were randomized into control and treated groups. In all studies, tumor volume (measured by caliper), animal body weight, and condition were
monitored at least twice weekly. 22Rv1 xenograft RNA was after 28 days of treatment of 20 mg/kg of CCS1477 (26 for the vehicle) and that the CP50cs were short term treated for 8 days and
long term was until they reached 300% of their original growth. Tumor samples were collected for analyses of pharmacodynamics biomarkers. All the mice used in this study were male. No mice
lost more than 5% of their body weight throughout the duration of the study. All mouse work was carried out in accordance with the Institute of Cancer Research (ICR) guidelines, including
approval by the ICR Animal Welfare and Ethical Review Body, and with the UK Animals (Scientific Procedures) Act 1986. The CP50 PDX and CP50s PDX was derived from a metastatic lymph node
biopsy from a patient with CRPC who had received all standard-of-care therapies for prostate cancer as previously described [35]. PDE (PATIENT DERIVED EXPLANT) Fresh prostate cancer
specimens were obtained with written informed consent from men undergoing robotic radical prostatectomy at St Andrew’s Hospital, Adelaide, through the Australian Prostate Cancer BioResource.
Dissected tissue fragments were utilized as ex vivo _PDE_ cultures as previously described [53, 83, 124, 125]. University of Adelaide’s Institutional Review Board has reviewed this protocol
and deemed this research to follow federal regulations (Approval # HREC-2012-016). PDE cultures were treated with media containing CCS1477 (1 or 5 mM) or vehicle alone (DMSO) and harvested
after 48 h for immunohistochemistry (IHC) analyses of the proliferative marker, Ki67, and qPCR for gene expression analyses of HR target genes. For histological analysis, explants were
formalin-fixed and paraffin embedded. For RNA analysis, explants were stabilized in RNAlater® (Ambion, TX, USA) at 4 oC overnight and then stored at −80 oC. IMMUNOHISTOCHEMISTRY (IHC) For
PDEs, immunostaining of formalin-fixed paraffin-embedded sections (2–4 μm) was performed with the Bond RX automated stainer (Leica Biosystems, Germany). Antigen retrieval was 20 min at 100
oC using the Bond Epitope Retrieval Solution 2 (EDTA based buffer, pH 9). FFPE sections were stained with Ki67 primary antibody (Clone MIB-1, DAKO, Denmark) (1:200) and a goat anti-mouse IgG
biotinylated secondary antibody (DAKO, Denmark). using standard techniques previously described [126]. HOMOLOGOUS RECOMBINATION (HR) ACTIVITY ASSAY U2OS-DR-GFP cells are a modified
osteosarcoma cell line that were generated by Dr. Jasin and were utilized to assess HR activity as previously described [81, 82, 127]. Dr. Roger A. Greenberg (University of Pennsylvania)
provided these cells that were utilized in this study. These cell lines were transfected with siCBP or siP300 using Dharmafect 4 reagent following manufacturer’s instructions. 48 h post
transfection, ISce1 plasmid was transfected into cells to induce DNA breaks. Cells were treated with ATM inhibitor (KU-55933, Sigma-Aldrich SML1109) or CBP/p300 inhibitor (CCS1477,
CellCentric) for the last 16 h of the assay. Cells were harvested and GFP positive cells were quantified via flow cytometry. STATISTICS All experiments were performed in technical triplicate
with at least 3 biological replicates per condition. Data are displayed as mean ± standard error of the mean (SEM). Statistical significance (p < 0.05) was determined using Student’s
t-test, one-way ANOVA, and two-way ANOVA on GraphPad Prism Software as appropriate and indicated in applicable figure legends. For the analysis of patient biopsies, nuclear CBP and p300
protein levels were reported as median values with IQRs. For paired and/or same PCa patient expression studies, the Wilcoxon matched pair signed rank test was used to compare differences in
protein expression levels. The correlation between nuclear CBP and p300 protein expression was determined using Spearman correlation. Overall survival was defined as time from diagnosis
(defined above) to date of death or last follow-up/contact. Patient outcomes were compared by nuclear CBP and p300 protein expression (H-score) at diagnosis; median overall survival and
median time to CRPC were estimated using the Kaplan–Meier method, and respective hazard ratios were obtained by Cox regression. DISCLAIMER The contents of this publication are the sole
responsibility of the author(s) and do not necessarily reflect the views, opinions, or policies of Uniformed Services University of the Health Sciences (USUHS), The Henry M. Jackson
Foundation for the Advancement of Military Medicine, Inc., the Department of Defense (DoD), the Departments of the Army, Navy, or Air Force. Mention of trade names, commercial products, or
organizations does not imply endorsement by the U.S. Government. DATA AVAILABILITY The datasets generated during the current study have been deposited in public repositories. RNA-Seq data
will be deposited in the NCBI GEO with the accession code upon publication. REFERENCES * Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA A Cancer J Clin.
2009;59:225–49. Google Scholar * Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA A Cancer J Clin. 2023;73:17–48. Google Scholar * Shafi AA, Yen AE, Weigel NL. Androgen
receptors in hormone-dependent and castration-resistant prostate cancer. Pharm Ther. 2013;140:223–38. CAS Google Scholar * Augello MA, Den RB, Knudsen KE. AR function in promoting
metastatic prostate cancer. Cancer Metastasis Rev. 2014;33:399–411. PubMed PubMed Central CAS Google Scholar * Balk SP, Knudsen KE. AR, the cell cycle, and prostate cancer. Nucl Recept
Signal. 2008;6:e001. PubMed PubMed Central Google Scholar * Burd CJ, Morey LM, Knudsen KE. Androgen receptor corepressors and prostate cancer. Endocr Relat Cancer. 2006;13:979–94. PubMed
CAS Google Scholar * Schrecengost R, Knudsen KE. Molecular pathogenesis and progression of prostate cancer. Semin Oncol. 2013;40:244–58. PubMed PubMed Central CAS Google Scholar *
Lytton B. Prostate cancer: a brief history and the discovery of hormonal ablation treatment. J Urol. 2001;165:1859–62. PubMed CAS Google Scholar * Knudsen KE, Scher HI. Starving the
addiction: new opportunities for durable suppression of AR signaling in prostate cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2009;15:4792–8. CAS Google Scholar * Sowery RD, So AI,
Gleave ME. Therapeutic options in advanced prostate cancer: present and future. Curr Urol Rep. 2007;8:53–59. PubMed Google Scholar * Omlin A, de Bono JS. Therapeutic options for advanced
prostate cancer: 2011 update. Curr Urol Rep. 2012;13:170–8. PubMed Google Scholar * Jacob A, Raj R, Allison DB, Myint ZW. Androgen receptor signaling in prostate cancer and therapeutic
strategies. Cancers. 2021;13:5417. * Reiss AB, Saeedullah U, Grossfeld DJ, Glass AD, Pinkhasov A, Katz AE. Prostate cancer treatment and the relationship of androgen deprivation therapy to
cognitive function. Clin Transl Oncol. 2022;24:733–41. PubMed CAS Google Scholar * Achard V, Putora PM, Omlin A, Zilli T, Fischer S. Metastatic prostate cancer: treatment options.
Oncology. 2022;100:48–59. PubMed CAS Google Scholar * Hewish M, Lord CJ, Martin SA, Cunningham D, Ashworth A. Mismatch repair deficient colorectal cancer in the era of personalized
treatment. Nat Rev Clin Oncol. 2010;7:197–208. PubMed Google Scholar * Hugh J, Hanson J, Cheang MC, Nielsen TO, Perou CM, Dumontet C, et al. Breast cancer subtypes and response to
docetaxel in node-positive breast cancer: use of an immunohistochemical definition in the BCIRG 001 trial. J Clin Oncol Off J Am Soc Clin Oncol. 2009;27:1168–76. CAS Google Scholar * Kim
C, Paik S. Gene-expression-based prognostic assays for breast cancer. Nat Rev Clin Oncol. 2010;7:340–7. PubMed CAS Google Scholar * Peppercorn J, Perou CM, Carey LA. Molecular subtypes in
breast cancer evaluation and management: divide and conquer. Cancer Investig. 2008;26:1–10. CAS Google Scholar * Zhao SG, Chang SL, Erho N, Yu M, Lehrer J, Alshalalfa M, et al.
Associations of luminal and basal subtyping of prostate cancer with prognosis and response to androgen deprivation therapy. JAMA Oncol. 2017;3:1663–72. PubMed PubMed Central Google Scholar
* Schiewer MJ, Knudsen KE. Linking DNA damage and hormone signaling pathways in cancer. Trends Endocrinol Metab. 2016;27:216–25. PubMed PubMed Central CAS Google Scholar * Wang Q, Li
W, Zhang Y, Yuan X, Xu K, Yu J, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–56. PubMed PubMed Central
CAS Google Scholar * Anantharaman A, Friedlander TW. Targeting the androgen receptor in metastatic castrate-resistant prostate cancer: a review. Urol Oncol. 2016;34:356–67. PubMed CAS
Google Scholar * Jadhavar PS, Ramachandran SA, Riquelme E, Gupta A, Quinn KP, Shivakumar D, et al. Targeting prostate cancer with compounds possessing dual activity as androgen receptor
antagonists and HDAC6 inhibitors. Bioorg Med Chem Lett. 2016;26:5222–8. PubMed CAS Google Scholar * Li D, Zhou W, Pang J, Tang Q, Zhong B, Shen C, et al. A magic drug target: androgen
receptor. Med Res Rev. 2019;39:1485–514. PubMed CAS Google Scholar * Schmidt AL, Tucker MD, Bakouny Z, Labaki C, Hsu CY, Shyr Y, et al. Association between androgen deprivation therapy
and mortality among patients with prostate cancer and COVID-19. JAMA Netw Open. 2021;4:e2134330. PubMed PubMed Central Google Scholar * Shah H, Vaishampayan U. Therapy of advanced
prostate cancer: targeting the androgen receptor axis in earlier lines of treatment. Target Oncol. 2018;13:679–89. PubMed PubMed Central Google Scholar * Bluemn EG, Coleman IM, Lucas JM,
Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell. 2017;32:474–489.e476. PubMed PubMed
Central CAS Google Scholar * Coutinho I, Day TK, Tilley WD, Selth LA. Androgen receptor signaling in castration-resistant prostate cancer: a lesson in persistence. Endocr Relat Cancer.
2016;23:T179–T197. PubMed CAS Google Scholar * Pal SK, Patel J, He M, Foulk B, Kraft K, Smirnov DA, et al. Identification of mechanisms of resistance to treatment with abiraterone acetate
or enzalutamide in patients with castration-resistant prostate cancer (CRPC). Cancer. 2018;124:1216–24. PubMed CAS Google Scholar * Schweizer MT, Yu EY. Persistent androgen receptor
addiction in castration-resistant prostate cancer. J Hematol Oncol. 2015;8:128. PubMed PubMed Central Google Scholar * Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance
to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15:701–11. PubMed PubMed Central CAS Google Scholar * Waddell AR, Huang H, Liao D CBP/p300: Critical
co-activators for nuclear steroid hormone receptors and emerging therapeutic targets in prostate and breast cancers. Cancers. 2021;13:2872. * Zou LJ, Xiang QP, Xue XQ, Zhang C, Li CC, Wang
C, et al. Y08197 is a novel and selective CBP/EP300 bromodomain inhibitor for the treatment of prostate cancer. Acta Pharm Sin. 2019;40:1436–47. CAS Google Scholar * Heemers HV, Debes JD,
Tindall DJ. The role of the transcriptional coactivator p300 in prostate cancer progression. Adv Exp Med Biol. 2008;617:535–40. PubMed PubMed Central Google Scholar * Welti J, Sharp A,
Brooks N, Yuan W, McNair C, Chand SN, et al. Targeting the p300/CBP axis in lethal prostate cancer. Cancer Discov. 2021;11:1118–37. PubMed PubMed Central CAS Google Scholar * He ZX, Wei
BF, Zhang X, Gong YP, Ma LY, Zhao W. Current development of CBP/p300 inhibitors in the last decade. Eur J Med Chem. 2021;209:112861. PubMed CAS Google Scholar * Jin L, Garcia J, Chan E,
de la Cruz C, Segal E, Merchant M, et al. Therapeutic targeting of the CBP/p300 bromodomain blocks the growth of castration-resistant prostate cancer. Cancer Res. 2017;77:5564–75. PubMed
CAS Google Scholar * Pan W, Zhang Z, Kimball H, Qu F, Berlind K, Stopsack KH, et al. Abiraterone acetate induces CREB1 phosphorylation and enhances the function of the CBP-p300 complex,
leading to resistance in prostate cancer cells. Clin Cancer Res Off J Am Assoc Cancer Res. 2021;27:2087–99. CAS Google Scholar * Yan Y, Ma J, Wang D, Lin D, Pang X, Wang S, et al. The
novel BET-CBP/p300 dual inhibitor NEO2734 is active in SPOP mutant and wild-type prostate cancer. EMBO Mol Med. 2019;11:e10659. PubMed PubMed Central CAS Google Scholar * Zhong J, Ding
L, Bohrer LR, Pan Y, Liu P, Zhang J, et al. p300 acetyltransferase regulates androgen receptor degradation and PTEN-deficient prostate tumorigenesis. Cancer Res. 2014;74:1870–80. PubMed
PubMed Central CAS Google Scholar * Aarnisalo P, Palvimo JJ, Janne OA. CREB-binding protein in androgen receptor-mediated signaling. Proc Natl Acad Sci USA. 1998;95:2122–7. PubMed PubMed
Central CAS Google Scholar * Comuzzi B, Lambrinidis L, Rogatsch H, Godoy-Tundidor S, Knezevic N, Krhen I, et al. The transcriptional co-activator cAMP response element-binding
protein-binding protein is expressed in prostate cancer and enhances androgen- and anti-androgen-induced androgen receptor function. Am J Pathol. 2003;162:233–41. PubMed PubMed Central CAS
Google Scholar * Debes JD, Schmidt LJ, Huang H, Tindall DJ. p300 mediates androgen-independent transactivation of the androgen receptor by interleukin 6. Cancer Res. 2002;62:5632–6.
PubMed CAS Google Scholar * Debes JD, Sebo TJ, Lohse CM, Murphy LM, Haugen DA, Tindall DJ. p300 in prostate cancer proliferation and progression. Cancer Res. 2003;63:7638–40. PubMed CAS
Google Scholar * Fronsdal K, Engedal N, Slagsvold T, Saatcioglu F. CREB binding protein is a coactivator for the androgen receptor and mediates cross-talk with AP-1. J Biol Chem.
1998;273:31853–9. PubMed CAS Google Scholar * Gruber M, Ferrone L, Puhr M, Santer FR, Furlan T, Eder IE, et al. p300 is upregulated by docetaxel and is a target in chemoresistant prostate
cancer. Endocr Relat Cancer. 2020;27:187–98. PubMed PubMed Central CAS Google Scholar * Ianculescu I, Wu DY, Siegmund KD, Stallcup MR. Selective roles for cAMP response element-binding
protein binding protein and p300 protein as coregulators for androgen-regulated gene expression in advanced prostate cancer cells. J Biol Chem. 2012;287:4000–13. PubMed CAS Google Scholar
* Lee YH, Hong SW, Jun W, Cho HY, Kim HC, Jung MG, et al. Anti-histone acetyltransferase activity from allspice extracts inhibits androgen receptor-dependent prostate cancer cell growth.
Biosci Biotechnol Biochem. 2007;71:2712–9. PubMed CAS Google Scholar * Santer FR, Hoschele PP, Oh SJ, Erb HH, Bouchal J, Cavarretta IT, et al. Inhibition of the acetyltransferases p300
and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol Cancer Ther. 2011;10:1644–55. PubMed CAS Google Scholar * Spriano
F, Gaudio E, Cascione L, Tarantelli C, Melle F, Motta G, et al. Antitumor activity of the dual BET and CBP/EP300 inhibitor NEO2734. Blood Adv. 2020;4:4124–35. PubMed PubMed Central CAS
Google Scholar * Goodwin JF, Knudsen KE. Beyond DNA repair: DNA-PK function in cancer. Cancer Discov. 2014;4:1126–39. PubMed PubMed Central CAS Google Scholar * Goodwin JF, Schiewer MJ,
Dean JL, Schrecengost RS, de Leeuw R, Han S, et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013;3:1254–71. PubMed CAS Google Scholar *
Goodwin JF, Kothari V, Drake JM, Zhao S, Dylgjeri E, Dean JL, et al. DNA-PKcs-mediated transcriptional regulation drives prostate cancer progression and metastasis. Cancer Cell.
2015;28:97–113. PubMed PubMed Central CAS Google Scholar * Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, et al. Androgen receptor signaling regulates DNA repair in
prostate cancers. Cancer Discov. 2013;3:1245–53. PubMed PubMed Central CAS Google Scholar * Ogiwara H, Kohno T. CBP and p300 histone acetyltransferases contribute to homologous
recombination by transcriptionally activating the BRCA1 and RAD51 genes. PLoS One. 2012;7:e52810. PubMed PubMed Central CAS Google Scholar * Raisner R, Kharbanda S, Jin L, Jeng E, Chan
E, Merchant M, et al. Enhancer activity requires CBP/P300 bromodomain-dependent histone H3K27 acetylation. Cell Rep. 2018;24:1722–9. PubMed CAS Google Scholar * Holmqvist PH, Mannervik M.
Genomic occupancy of the transcriptional co-activators p300 and CBP. Transcription. 2013;4:18–23. PubMed Google Scholar * Martire S, Nguyen J, Sundaresan A, Banaszynski LA. Differential
contribution of p300 and CBP to regulatory element acetylation in mESCs. BMC Mol Cell Biol. 2020;21:55. PubMed PubMed Central CAS Google Scholar * Cai C, Hsieh CL, Omwancha J, Zheng Z,
Chen SY, Baert JL, et al. ETV1 is a novel androgen receptor-regulated gene that mediates prostate cancer cell invasion. Mol Endocrinol. 2007;21:1835–46. PubMed CAS Google Scholar *
Grabowska MM, Elliott AD, DeGraff DJ, Anderson PD, Anumanthan G, Yamashita H, et al. NFI transcription factors interact with FOXA1 to regulate prostate-specific gene expression. Mol
Endocrinol. 2014;28:949–64. PubMed PubMed Central Google Scholar * Rahim S, Beauchamp EM, Kong Y, Brown ML, Toretsky JA, Uren A. YK-4-279 inhibits ERG and ETV1 mediated prostate cancer
cell invasion. PLoS One. 2011;6:e19343. PubMed PubMed Central CAS Google Scholar * Sankpal UT, Goodison S, Abdelrahim M, Basha R. Targeting Sp1 transcription factors in prostate cancer
therapy. Med Chem. 2011;7:518–25. PubMed CAS Google Scholar * Sun Q, Yu X, Degraff DJ, Matusik RJ. Upstream stimulatory factor 2, a novel FoxA1-interacting protein, is involved in
prostate-specific gene expression. Mol Endocrinol. 2009;23:2038–47. PubMed PubMed Central CAS Google Scholar * Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al.
Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. PubMed CAS Google Scholar * Zhu Z, Luo L, Xiang Q, Wang J, Liu Y, Deng Y, et
al. MiRNA-671-5p Promotes prostate cancer development and metastasis by targeting NFIA/CRYAB axis. Cell Death Dis. 2020;11:949. PubMed PubMed Central CAS Google Scholar * Ikeuchi W,
Wakita Y, Zhang G, Li C, Itakura K, Yamakawa T. AT-rich interaction domain 5A regulates the transcription of interleukin-6 gene in prostate cancer cells. Prostate. 2022;82:97–106. PubMed
CAS Google Scholar * Ghadersohi A, Odunsi K, Lele S, Collins Y, Greco WR, Winston J, et al. Prostate derived Ets transcription factor shows better tumor-association than other
cancer-associated molecules. Oncol Rep. 2004;11:453–8. PubMed CAS Google Scholar * Mahajan N. Signatures of prostate-derived Ets factor (PDEF) in cancer. Tumour Biol. 2016;37:14335–40.
PubMed CAS Google Scholar * Mitas M, Mikhitarian K, Hoover L, Lockett MA, Kelley L, Hill A, et al. Prostate-specific Ets (PSE) factor: a novel marker for detection of metastatic breast
cancer in axillary lymph nodes. Br J Cancer. 2002;86:899–904. PubMed PubMed Central CAS Google Scholar * Scimeca M, Montanaro M, Bonfiglio R, Anemona L, Agro EF, Asimakopoulos AD, et al.
The ETS homologous factor (EHF) represents a useful immunohistochemical marker for predicting prostate cancer metastasis. Diagnostics. 2022;12:800. * Halabi S, Guo S, Park JJ, Nanus DM,
George DJ, Antonarakis ES, et al. The impact of circulating tumor cell HOXB13 RNA detection in men with metastatic castration-resistant prostate cancer (mCRPC) treated with abiraterone or
enzalutamide. Clin Cancer Res Off J Am Assoc Cancer Res. 2024;30:1152–9. CAS Google Scholar * Heise M, Jarzemski P, Bak A, Junkiert-Czarnecka A, Pilarska-Deltow M, Haus O. G84E germline
mutation in HOXB13 gene is associated with increased prostate cancer risk in Polish men. Pol J Pathol. 2019;70:127–33. PubMed Google Scholar * Jung C, Kim RS, Lee SJ, Wang C, Jeng MH.
HOXB13 homeodomain protein suppresses the growth of prostate cancer cells by the negative regulation of T-cell factor 4. Cancer Res. 2004;64:3046–51. PubMed CAS Google Scholar * Jung C,
Kim RS, Zhang HJ, Lee SJ, Jeng MH. HOXB13 induces growth suppression of prostate cancer cells as a repressor of hormone-activated androgen receptor signaling. Cancer Res. 2004;64:9185–92.
PubMed CAS Google Scholar * Patel RA, Sayar E, Coleman I, Roudier MP, Hanratty B, Low JY, et al. Characterization of HOXB13 expression patterns in localized and metastatic
castration-resistant prostate cancer. J Pathol. 2024;262:105–20. PubMed CAS Google Scholar * Trendowski MR, Sample C, Baird T, Sadeghpour A, Moon D, Ruterbusch JJ, et al. Germline
variants in DNA damage repair genes and HOXB13 among black patients with early-onset prostate cancer. JCO Precis Oncol. 2022;6:e2200460. PubMed PubMed Central Google Scholar * Anno YN,
Myslinski E, Ngondo-Mbongo RP, Krol A, Poch O, Lecompte O, et al. Genome-wide evidence for an essential role of the human Staf/ZNF143 transcription factor in bidirectional transcription.
Nucleic Acids Res. 2011;39:3116–27. PubMed CAS Google Scholar * Izumi H, Wakasugi T, Shimajiri S, Tanimoto A, Sasaguri Y, Kashiwagi E, et al. Role of ZNF143 in tumor growth through
transcriptional regulation of DNA replication and cell-cycle-associated genes. Cancer Sci. 2010;101:2538–45. PubMed PubMed Central CAS Google Scholar * Velho PI, Bastos DA, Antonarakis
ES. New approaches to targeting the androgen receptor pathway in prostate cancer. Clin Adv Hematol Oncol. 2021;19:228–40. PubMed Google Scholar * Shafi AA, McNair CM, McCann JJ, Alshalalfa
M, Shostak A, Severson TM, et al. The circadian cryptochrome, CRY1, is a pro-tumorigenic factor that rhythmically modulates DNA repair. Nat Commun. 2021;12:401. PubMed PubMed Central CAS
Google Scholar * Johnson RD, Jasin M. Double-strand-break-induced homologous recombination in mammalian cells. Biochem Soc Trans. 2001;29:196–201. PubMed CAS Google Scholar * Vriend
LE, Jasin M, Krawczyk PM. Assaying break and nick-induced homologous recombination in mammalian cells using the DR-GFP reporter and Cas9 nucleases. Methods Enzymol. 2014;546:175–91. PubMed
PubMed Central CAS Google Scholar * Shafi AA, Schiewer MJ, de Leeuw R, Dylgjeri E, McCue PA, Shah N, et al. Patient-derived models reveal impact of the tumor microenvironment on
therapeutic response. Eur Urol Oncol. 2018;1:325–37. PubMed PubMed Central Google Scholar * Ramos YF, Hestand MS, Verlaan M, Krabbendam E, Ariyurek Y, van Galen M, et al. Genome-wide
assessment of differential roles for p300 and CBP in transcription regulation. Nucleic Acids Res. 2010;38:5396–408. PubMed PubMed Central CAS Google Scholar * Lasko LM, Jakob CG, Edalji
RP, Qiu W, Montgomery D, Digiammarino EL, et al. Author correction: discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature. 2018;558:E1. PubMed
CAS Google Scholar * Yuan ZM, Huang Y, Ishiko T, Nakada S, Utsugisawa T, Shioya H, et al. Role for p300 in stabilization of p53 in the response to DNA damage. J Biol Chem. 1999;274:1883–6.
PubMed CAS Google Scholar * Iyer NG, Chin SF, Ozdag H, Daigo Y, Hu DE, Cariati M, et al. p300 regulates p53-dependent apoptosis after DNA damage in colorectal cancer cells by modulation
of PUMA/p21 levels. Proc Natl Acad Sci USA. 2004;101:7386–91. PubMed PubMed Central CAS Google Scholar * Ansari MSZ, Stagni V, Iuzzolino A, Rotili D, Mai A, Del Bufalo D, et al.
Pharmacological targeting of CBP/p300 drives a redox/autophagy axis leading to senescence-induced growth arrest in non-small cell lung cancer cells. Cancer Gene Ther. 2023;30:124–36. PubMed
CAS Google Scholar * Schiewer MJ, Mandigo AC, Gordon N, Huang F, Gaur S, de Leeuw R, et al. PARP-1 regulates DNA repair factor availability. EMBO Mol Med. 2018;10:e8816. * Hasterok S,
Scott TG, Roller DG, Spencer A, Dutta AB, Sathyan KM, et al. The androgen receptor does not directly regulate the transcription of DNA damage response genes. Mol Cancer Res. 2023;21:1329–41.
PubMed PubMed Central CAS Google Scholar * Yao W, Wang T, Huang F. p300/CBP as a key nutritional sensor for hepatic energy homeostasis and liver fibrosis. Biomed Res Int.
2018;2018:8168791. PubMed PubMed Central Google Scholar * Cai LY, Chen SJ, Xiao SH, Sun QJ, Ding CH, Zheng BN, et al. Targeting p300/CBP attenuates hepatocellular carcinoma progression
through epigenetic regulation of metabolism. Cancer Res. 2021;81:860–72. PubMed CAS Google Scholar * Buschmann T, Adler V, Matusevich E, Fuchs SY, Ronai Z. p53 phosphorylation and
association with murine double minute 2, c-Jun NH2-terminal kinase, p14ARF, and p300/CBP during the cell cycle and after exposure to ultraviolet irradiation. Cancer Res. 2000;60:896–900.
PubMed CAS Google Scholar * Somasundaram K, El-Deiry WS. Inhibition of p53-mediated transactivation and cell cycle arrest by E1A through its p300/CBP-interacting region. Oncogene.
1997;14:1047–57. PubMed CAS Google Scholar * Turnell AS, Stewart GS, Grand RJ, Rookes SM, Martin A, Yamano H, et al. The APC/C and CBP/p300 cooperate to regulate transcription and
cell-cycle progression. Nature. 2005;438:690–5. PubMed CAS Google Scholar * Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, et al. Dual roles of PARP-1 promote cancer
growth and progression. Cancer Discov. 2012;2:1134–49. PubMed PubMed Central CAS Google Scholar * Schiewer MJ, Knudsen KE. Transcriptional roles of PARP1 in cancer. Mol Cancer Res.
2014;12:1069–80. PubMed PubMed Central CAS Google Scholar * Steffen JD, Tholey RM, Langelier MF, Planck JL, Schiewer MJ, Lal S, et al. Targeting PARP-1 allosteric regulation offers
therapeutic potential against cancer. Cancer Res. 2014;74:31–37. PubMed CAS Google Scholar * Chakraborty G, Armenia J, Mazzu YZ, Nandakumar S, Stopsack KH, Atiq MO, et al. Significance of
BRCA2 and RB1 co-loss in aggressive prostate cancer progression. Clin Cancer Res Off J Am Assoc Cancer Res. 2020;26:2047–64. CAS Google Scholar * Pham MT, Gupta A, Gupta H, Vaghasia A,
Skaist A, Garrison MA, et al. Identifying phased mutations and complex rearrangements in human prostate cancer cell lines through linked-read whole-genome sequencing. Mol Cancer Res.
2022;20:1013–20. PubMed PubMed Central CAS Google Scholar * Lotan TL, Kaur HB, Salles DC, Murali S, Schaeffer EM, Lanchbury JS, et al. Homologous recombination deficiency (HRD) score in
germline BRCA2- versus ATM-altered prostate cancer. Mod Pathol. 2021;34:1185–93. PubMed PubMed Central CAS Google Scholar * Zhu S, Zhao J, Nie L, Yin W, Zhang Y, Zhao F, et al.
Homologous recombination deficiency (HRD) score in aggressive prostatic adenocarcinoma with or without intraductal carcinoma of the prostate (IDC-P). BMC Med. 2022;20:237. PubMed PubMed
Central CAS Google Scholar * Beije N, Abida W, Antonarakis ES, Castro E, de Wit R, Fizazi K, et al. PARP inhibitors for prostate cancer: tangled up in PROfound and PROpel (and TALAPRO-2)
blues. Eur Urol. 2023;84:253–6. PubMed CAS Google Scholar * Gaur AS. PROpel trial: are PARP inhibitors ready to become the first-line treatment for mCRPC? Indian J Urol. 2022;38:319–20.
PubMed PubMed Central Google Scholar * Gu L, Du N, Jin Q, Li S, Xie L, Mo J, et al. Magnitude of benefit of the addition of poly ADP-ribose polymerase (PARP) inhibitors to therapy for
malignant tumor: a meta-analysis. Crit Rev Oncol Hematol. 2020;147:102888. PubMed Google Scholar * Carreira S, Porta N, Arce-Gallego S, Seed G, Llop-Guevara A, Bianchini D, et al.
Biomarkers associating with PARP inhibitor benefit in prostate cancer in the TOPARP-B Trial. Cancer Discov. 2021;11:2812–27. PubMed PubMed Central CAS Google Scholar * Mateo J, Porta N,
Bianchini D, McGovern U, Elliott T, Jones R, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre,
open-label, randomised, phase 2 trial. Lancet Oncol. 2020;21:162–74. PubMed PubMed Central CAS Google Scholar * Abida W, Campbell D, Patnaik A, Shapiro JD, Sautois B, Vogelzang NJ, et
al. Non-BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: analysis from the phase II TRITON2 study.
Clin Cancer Res Off J Am Assoc Cancer Res. 2020;26:2487–96. CAS Google Scholar * Helleday T. PARP inhibitor receives FDA breakthrough therapy designation in castration resistant prostate
cancer: beyond germline BRCA mutations. Ann Oncol. 2016;27:755–7. PubMed CAS Google Scholar * Maiorano BA, De Giorgi U, Verzoni E, Maiello E, Procopio G, Conteduca V, et al. Hematological
toxicity of PARP inhibitors in metastatic prostate cancer patients with mutations of BRCA or HRR genes: a systematic review and safety meta-analysis. Target Oncol. 2024;19:1–11. PubMed
Google Scholar * Shapiro GI, Barry SM. Combining PARP inhibition and immunotherapy in BRCA-associated cancers. Cancer Treat Res. 2023;186:207–21. PubMed Google Scholar * Stopsack KH.
Efficacy of PARP inhibition in metastatic castration-resistant prostate cancer is very different with non-BRCA DNA repair alterations: reconstructing prespecified endpoints for cohort B from
the phase 3 PROfound trial of olaparib. Eur Urol. 2021;79:442–5. PubMed CAS Google Scholar * Nicosia L, Spencer GJ, Brooks N, Amaral FMR, Basma NJ, Chadwick JA, et al. Therapeutic
targeting of EP300/CBP by bromodomain inhibition in hematologic malignancies. Cancer Cell. 2023;41:2136–2153 e2113. PubMed CAS Google Scholar * Chang Q, Li J, Deng Y, Zhou R, Wang B, Wang
Y, et al. Discovery of Novel PROTAC Degraders of p300/CBP as Potential Therapeutics for Hepatocellular Carcinoma. J Med Chem. 2024;67:2466–86. PubMed CAS Google Scholar * Chen Z, Wang M,
Wu D, Zhao L, Metwally H, Jiang W, et al. Discovery of CBPD-409 as a highly potent, selective, and orally efficacious CBP/p300 PROTAC degrader for the treatment of advanced prostate cancer.
J Med Chem. 2024;67:5371–72. * Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci USA.
2019;116:11428–36. PubMed PubMed Central CAS Google Scholar * Fenor de la Maza MD, Chandran K, Rekowski J, Shui IM, Gurel B, Cross E, et al. Immune biomarkers in metastatic
castration-resistant prostate cancer. Eur Urol Oncol. 2022;5:659–67. PubMed Google Scholar * Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal
RNA-seq aligner. Bioinformatics. 2013;29:15–21. PubMed CAS Google Scholar * Dobin A, Gingeras TR. Optimizing RNA-seq mapping with STAR. Methods Mol Biol. 2016;1415:245–62. PubMed CAS
Google Scholar * Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30. PubMed CAS
Google Scholar * Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. PubMed PubMed Central Google
Scholar * Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 2005;102:15545–50. PubMed PubMed Central CAS Google Scholar * de Leeuw R, McNair C, Schiewer MJ, Neupane NP, Brand LJ, Augello MA, et al. MAPK reliance
via acquired CDK4/6 inhibitor resistance in cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2018;24:4201–14. Google Scholar * Centenera MM, Hickey TE, Jindal S, Ryan NK, Ravindranathan
P, Mohammed H, et al. A patient-derived explant (PDE) model of hormone-dependent cancer. Mol Oncol. 2018;12:1608–22. PubMed PubMed Central Google Scholar * de Leeuw R, Berman-Booty LD,
Schiewer MJ, Ciment SJ, Den RB, Dicker AP, et al. Novel actions of next-generation taxanes benefit advanced stages of prostate cancer. Clin Cancer Res Off J Am Assoc Cancer Res.
2015;21:795–807. Google Scholar * Centenera MM, Vincent AD, Moldovan M, Lin HM, Lynn DJ, Horvath LG, et al. Harnessing the heterogeneity of prostate cancer for target discovery using
patient-derived explants. Cancers. 2022;14:1708. * Jasin M, de Villiers J, Weber F, Schaffner W. High frequency of homologous recombination in mammalian cells between endogenous and
introduced SV40 genomes. Cell. 1985;43:695–703. PubMed CAS Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by a Young Investigator Award and Challenge Award
from the Prostate Cancer Foundation (to AAS, AS, and KEK), NCI R01-CA182569 (KEK), the Sidney Kimmel Cancer Center (SKCC) Support Grant (5P30CA056036), CPDR-CORE Funds (AAS, XAS), Wellcome
Trust Clinical Research Career Development Fellowship (AS) and the Cancer Genomics and Translational Research/Pathology core services at SKCC. LMB is supported by the South Australian
Immunogenomics Cancer Institute at the University of Adelaide and Cancer Australia (ID#1138766). Additionally, we would like to thank Dr. R. Greenberg (University of Pennsylvania) for
providing the U2OS-DR-GFP cells. The authors thank the study participants, urologists, nurses, histopathologists, and coordinators of the Australian Prostate Cancer Bioresource that provided
specimens for this research. The collaborators played a role in the design of the study; data collection; analysis; interpretation; and review and approval of the manuscript. Lastly, we
would like to thank former Knudsen lab members and current CPDR colleagues for providing suggestions and support for this study. AUTHOR INFORMATION Author notes * These authors contributed
equally: Sumaira Sardar, Christopher M. McNair. AUTHORS AND AFFILIATIONS * Center for Prostate Disease Research, Murtha Cancer Center Research Program, Department of Surgery, Uniformed
Services University of the Health Sciences, Bethesda, MD, USA Sumaira Sardar, Lakshmi Ravindranath, Elise G. DeArment, Thomas Janas, Xiaofeng A. Su & Ayesha A. Shafi * The Henry M.
Jackson Foundation for the Advancement of Military Medicine Inc., Bethesda, MD, USA Sumaira Sardar, Lakshmi Ravindranath, Elise G. DeArment, Thomas Janas, Xiaofeng A. Su & Ayesha A.
Shafi * Sidney Kimmel Cancer Center, Thomas Jefferson University, Philadelphia, PA, USA Christopher M. McNair, Saswati N. Chand, Liam A. Knudsen & Matthew J. Schiewer * The Institute of
Cancer Research, London, United Kingdom Wei Yuan, Denisa Bogdan, Jon Welti, Adam Sharp & Johann S. de Bono * The Royal Marsden Hospital, London, United Kingdom Adam Sharp & Johann S.
de Bono * South Australian Immunogenomics Cancer Institute, The University of Adelaide, Adelaide, SA, Australia Natalie K. Ryan & Lisa M. Butler * South Australian Health and Medical
Research Institute, Adelaide, SA, Australia Natalie K. Ryan & Lisa M. Butler * David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge,
MA, USA Xiaofeng A. Su * CellCentric Ltd., Cambridge, United Kingdom Kris Frese, Nigel Brooks & Neil Pegg * The American Cancer Society, Philadelphia, PA, USA Karen E. Knudsen Authors *
Sumaira Sardar View author publications You can also search for this author inPubMed Google Scholar * Christopher M. McNair View author publications You can also search for this author
inPubMed Google Scholar * Lakshmi Ravindranath View author publications You can also search for this author inPubMed Google Scholar * Saswati N. Chand View author publications You can also
search for this author inPubMed Google Scholar * Wei Yuan View author publications You can also search for this author inPubMed Google Scholar * Denisa Bogdan View author publications You
can also search for this author inPubMed Google Scholar * Jon Welti View author publications You can also search for this author inPubMed Google Scholar * Adam Sharp View author publications
You can also search for this author inPubMed Google Scholar * Natalie K. Ryan View author publications You can also search for this author inPubMed Google Scholar * Liam A. Knudsen View
author publications You can also search for this author inPubMed Google Scholar * Matthew J. Schiewer View author publications You can also search for this author inPubMed Google Scholar *
Elise G. DeArment View author publications You can also search for this author inPubMed Google Scholar * Thomas Janas View author publications You can also search for this author inPubMed
Google Scholar * Xiaofeng A. Su View author publications You can also search for this author inPubMed Google Scholar * Lisa M. Butler View author publications You can also search for this
author inPubMed Google Scholar * Johann S. de Bono View author publications You can also search for this author inPubMed Google Scholar * Kris Frese View author publications You can also
search for this author inPubMed Google Scholar * Nigel Brooks View author publications You can also search for this author inPubMed Google Scholar * Neil Pegg View author publications You
can also search for this author inPubMed Google Scholar * Karen E. Knudsen View author publications You can also search for this author inPubMed Google Scholar * Ayesha A. Shafi View author
publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Conceptualization, CMM, AS, XAS, LB, JB, KEK, and AAS; Methodology, CMM, KEK, and AAS; Software, CMM,
WY, and DB; Validation, SS CCM, LR, and AAS; Formal analysis, SS, CMM, SNC, WY, DB, JW, LAK, EGD, TJ, LB, and AAS; Investigation, AAS, MA, TMS, YZ, AB, NKR, and MJS; Resources, CMM, AS, MJS,
LB, JB, KF, NB, NP, KEK, and AAS; Writing, SS and AAS; Writing—Review and Editing, SS, CMM, AS, MJS, LB, JB, KF, NB, NP, KEK, and AAS; Visualization, SS, CMM, WY, DB, LB, JD, AAS; Funding
Acquisition, KEK, and AAS. CORRESPONDING AUTHOR Correspondence to Ayesha A. Shafi. ETHICS DECLARATIONS COMPETING INTERESTS The following are disclosures for authors on this manuscript: Karen
E. Knudsen is the CEO of American Cancer Society (ACS). Adam Sharp has received travel support from Sanofi, Roche-Genentech, and Nurix, and speaker honoraria from Astellas Pharma and Merck
Sharp & Dohme. He has served as an advisor to DE Shaw Research & CHARM Therapeutics and has been the CI/PI of industry-sponsored clinical trials. No disclosures for the other
authors. Neil Pegg, Nigel Brooks, and Kris Frese are employees and shareholders of CellCentric Ltd. Neil Pegg is also a board director and the inventor on CCS1477 patents. Johann S de Bono
reports grants from CellCentric during the conduct of the study; grants and personal fees from Daiichi Sankyo, AstraZeneca, Pfizer, Bayer Oncology, MSD, Merck Serono, Harpoon, and
Genentech/Roche, personal fees from Eisai and Constellation, and grants from Sun Pharma outside the submitted work; in addition, Johann S. de Bono has a patent for Abiraterone licensed and
with royalties paid from Janssen. No other disclosures were reported. ETHICAL APPROVAL The use of patient and clinical material was approved by the ethical committees from each of the
following institutes: the Sidney Kimmel Cancer Center at Thomas Jefferson University (Pennsylvania, USA), Institute of Cancer Research (ICR – London, United Kingdom), and the University of
Adelaide (Adelaide, Australia). ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURES AND LEGENDS RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0
International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s)
and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material
derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a
credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted
use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions
ABOUT THIS ARTICLE CITE THIS ARTICLE Sardar, S., McNair, C.M., Ravindranath, L. _et al._ AR coactivators, CBP/p300, are critical mediators of DNA repair in prostate cancer. _Oncogene_ 43,
3197–3213 (2024). https://doi.org/10.1038/s41388-024-03148-4 Download citation * Received: 24 June 2024 * Revised: 20 August 2024 * Accepted: 28 August 2024 * Published: 13 September 2024 *
Issue Date: 21 October 2024 * DOI: https://doi.org/10.1038/s41388-024-03148-4 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable
link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative