Proteinase 3 depletion attenuates leukemia by promoting myeloid differentiation
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Hematopoietic stem and progenitor cells (HSPCs) that have impaired differentiation can transform into leukemic blasts. However, the mechanism that controls differentiation remains
elusive. Here, we show that the genetic elimination of Proteinase 3 (PRTN3) in mice led to spontaneous myeloid differentiation. Mechanistically, our findings indicate that PRTN3 interacts
with the N-terminal of STAT3, serving as a negative regulator of STAT3-dependent myeloid differentiation. Specifically, PRTN3 promotes STAT3 ubiquitination and degradation, while
simultaneously reducing STAT3 phosphorylation and nuclear translocation during G-CSF-stimulated myeloid differentiation. Strikingly, pharmacological inhibition of STAT3 (Stattic) partially
counteracted the effects of PRTN3 deficiency on myeloid differentiation. Moreover, the deficiency of PRTN3 in primary AML blasts promotes the differentiation of those cells into functional
neutrophils capable of chemotaxis and phagocytosis, ultimately resulting in improved overall survival rates for recipients. These findings indicate PRTN3 exerts an inhibitory effect on
STAT3-dependent myeloid differentiation and could be a promising therapeutic target for the treatment of acute myeloid leukemia. SIMILAR CONTENT BEING VIEWED BY OTHERS MLKL PROMOTES CELLULAR
DIFFERENTIATION IN MYELOID LEUKEMIA BY FACILITATING THE RELEASE OF G-CSF Article Open access 02 June 2021 SELECTIVE ACTIVATION OF STAT3 AND STAT5 DICTATES THE FATE OF MYELOID PROGENITOR
CELLS Article Open access 28 July 2023 INTERLUKIN-4 WEAKENS RESISTANCE TO STRESS INJURY AND MEGAKARYOCYTIC DIFFERENTIATION OF HEMATOPOIETIC STEM CELLS BY INHIBITING _PSMD13_ EXPRESSION
Article Open access 31 August 2023 INTRODUCTION The differentiation of myeloid cells from progenitors is a vital process facilitated by hematopoietic cytokines, resulting in the development
of granulocytes and monocytes [1, 2]. These cells are crucial for the immune system’s defense against invading microorganisms, and their deficiencies can lead to severe pathological
conditions [3]. Arrest of myeloid differentiation can cause the accumulation of proliferative blasts, leading to the production of immature cells, as observed in myeloid leukemias [4].
However, differentiation therapy has shown success in treating acute myeloid leukemia (AML), particularly acute promyelocytic leukemia. This therapy promotes the maturation of AML cells and
facilitates the clearance of normal mature myeloid cells [5,6,7]. Therefore, identifying useful genetic targets that can induce hematopoietic maturation and differentiation has the potential
to be therapeutically significant across different AML subtypes. Proteinase 3 (PRTN3) is a neutral serine protease mainly found in neutrophils and monocytes, playing a significant role in
both the non-oxidative pathway of intracellular and extracellular pathogen destruction, as well as being a major component of neutrophil azurophilic granules [8, 9]. In addition to its role
in controlling apoptosis [9], PRTN3 has a proteolytic-independent microbicidal activity [10] and an elastase-like enzymatic activity [3]. Moreover, it promotes the degradation of
extracellular matrix and basement membrane proteins [11]. Although PRTN3 has been extensively studied in relation to its function in the maturation of myeloid cells, it is also crucial in
many other cellular functions, especially those involving hematopoietic stem and progenitor cells [9, 12,13,14]. Nevertheless, our understanding of the mechanism and the biological
significance of PRTN3 expressed in hematopoietic stem and progenitor cells to regulate myeloid differentiation is very limited. In this study, we aimed to investigate the effect of PRTN3
deficiency on spontaneous myeloid differentiation in mice and explore the potential of STAT3 inhibitors to counteract this effect. We explored the molecular mechanisms that regulate PRTN3 in
relation to STAT3-dependent myeloid differentiation both in vitro and in vivo. Furthermore, we demonstrated that PRTN3 deficiency protects against acute myeloid leukemia by promoting the
resumption of myeloid differentiation in mice and humans. Our findings reveal a novel pathway by which PRTN3 regulates myeloid differentiation by downregulating STAT3 expression, indicating
a promising new therapeutic target for the treatment of acute myeloid leukemia. RESULTS DEPLETION OF PRTN3 INDUCES MYELOID DIFFERENTIATION IN MICE To determine PRTN3’s expression in
hematopoietic cells, we conducted a data mining analysis using the Haemopedia RNA-seq data sets (https://www.haemosphere.org) to determine PRTN3’s expression in hematopoietic cells. The
findings revealed that PRTN3 is expressed throughout hematopoiesis, with the highest levels detected in multi-potential progenitor cells, restricted potential progenitor cells, and the
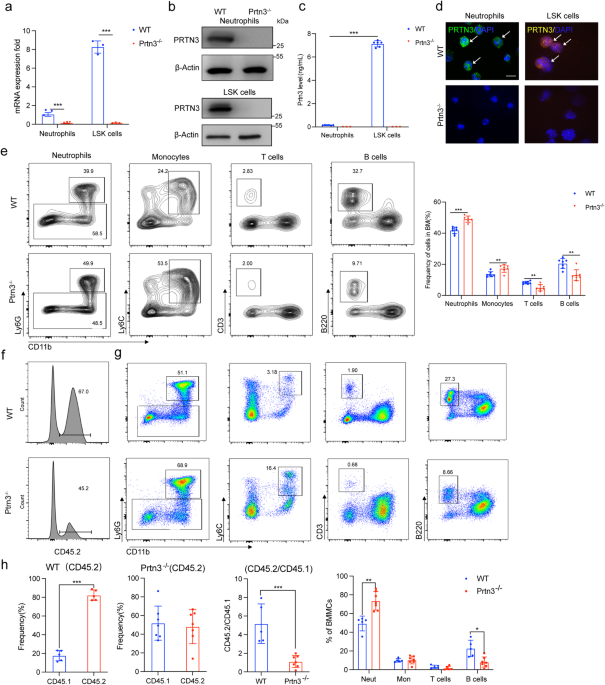
myeloid lineage (Fig. S1). To investigate whether PRTN3 deficiency affects myeloid differentiation, we generated global _Prtn3_ knockout mice (_Prtn3__−/−_) and confirmed _Prtn3_ deletion in
these mice using various methods, including RT-qPCR, Western blot, ELISA assay, and immunofluorescence staining (Fig. 1a-d). Harvesting bone marrow and flow cytometry to identify different
cell populations, we observed an increase in myeloid populations, neutrophil and monocyte, and a decrease in T and B cell populations in _Prtn3__−/−_ mice compared to their WT littermates
(Fig. 1e). Additionally, through routine analysis and flow cytometry, we found that _Prtn3__−/−_ mice exhibited a gradual exacerbation of spontaneous increases in myeloid cell populations,
specifically neutrophils and monocytes, and decreases populations of T cell, in bone marrow, spleen, and peripheral blood (Fig. S2a-d). We conducted a series of assays to assess T cell
functions, including the CFSE T cell proliferation assay, T cell activation bioassay, and T cell killing assay. Interestingly, our findings revealed that the proliferation, activation, and
anti-AML immunity of T cells were not affected in _Prtn3_−/− mice, compared to their WT littermates. (Fig. S3a-c). Next, we examined the number of LK and LSK cells and found a significant
increase in the percentage of these cells in _Prtn3__−/−_ mice, compared to WT mice from 24 weeks to 72 weeks (Fig. S4a, b). We further examined the percentage of GMP, CMP, MEP, MPP, ST-HSC,
and LT-HSC in both WT and _Prtn3__−/−_ mice throughout the aging process using flow cytometry. Our analysis revealed a significant increase in _Prtn3__−/−_ mice from 24 weeks to 72 weeks,
compared to WT mice, except for MEP (Fig. S4c, d). We also cultured LSK cells from mice of varying ages on metho-cult GM 3434 medium and found no change in colony-forming ability for all
colony types (G, M, GM, and GEMM) at stages 4, 8, 24, 48, and 72 weeks of isolated-primary cells for both WT and _Prtn3__−/−_ mice (Fig. S5). To confirm that PRTN3 affects myeloid
differentiation in LSK cells rather than downstream cells, we generated _Prtn3_ conditional knockout (_Prtn3__LyzKO_) mice by crossing _Prtn3__flox/flox_ mice with LysMCre mice. The
percentage of myeloid cells was analyzed through flow cytometry, and the results showed no difference between WT and _Prtn3__LyzKO_ mice in the percentage of neutrophils, monocytes, T cells,
and B cells in the bone marrow, spleen, and peripheral blood (Fig. S6). Thus, we conclude that _Prtn3_ deficiency in mice induces advanced spontaneous myeloid differentiation, and PRTN3 is
crucial for myeloid differentiation in LSK cells. To investigate whether the lack of _Prtn3_ promotes the differentiation of LSK cells into myeloid cells in mice receiving transplants, we
sorted 1 × 103 LSK cells from both WT and _Prtn3__−/−_ mice [C57BL6-Ly5.2/CD45.2(C57)] at 8, 48, and 72 weeks old, and mixed these cells with 5 × 105 bone marrow cells from
C57BL6-Ly5.2/CD45.1(B6) mice, which we then injected into C57BL6-Ly5.2/CD45.1(B6) mice that had undergone 9.5 Gy radiation. After 16 weeks post-transplant, we analyzed chimerism in the bone
marrow and spleen (Fig. S7a). Routine analysis of blood showed a higher percentage of neutrophils in the peripheral blood of _Prtn3__−/−_ mice than in WT mice at different ages (Fig. S7b,
Fig. S8a, and Fig. S9a). Flow cytometry analysis revealed that the ratio of CD45.2/CD45.1 was decreased, while the myeloid population was elevated in bone marrow (Fig. 1f, g, h, Fig. S8b,
Fig. S9b), spleen (Fig. S7c, Fig. S8c, Fig. S9c), and peripheral blood (Fig. S7d, Fig. S8d, Fig. S9d) of _Prtn3__−/−_ mice compared to WT mice at various ages. Furthermore, we observed an
increase in the percentage of various cell types, including LK cells, LSK cells, GMP, CMP, MEP, MPP, ST-HSC, and LT-HSC, in _Prtn3__−/−_ mice compared to WT mice, and this increase was
age-dependent (Fig. S7e, Fig. S8e, Fig. S9e). In addition, the immunofluorescence assay showed lower chimerism with donor-derived cells from _Prtn3__−/−_ mice in the recipient mice (Fig.
S7f, Fig. S8f, Fig. S9f). However, we did not observe a significant change in the number of colonies formed, including G, M, GM, and GEMM, when we cultured LSK cells from WT and _Prtn3__−/−_
mice of different ages (Fig. S7g, h, Fig. S8g, Fig. S9g). Our results suggest that PRTN3 plays a crucial role in the myeloid differentiation of LSK cells. PRTN3 DIRECTLY INTERACTS WITH
STAT3 To explore the molecular mechanism underlying myeloid differentiation and the potential factors that interact with PRTN3, we conducted immunoprecipitation (IP) assays and utilized
high-sensitivity mass spectrometry screening. Specifically, we investigated the binding of PRTN3 to potential proteins in HEK293T cells that overexpressed PRTN3 by transfecting an HA-tagged
PRTN3 plasmid and subsequently pulldown with the HA antibody. After screening over 5000 peptides, we identified one STAT family protein, STAT3, that may be linked to PRTN3 and the regulation
of myeloid differentiation (Fig. 2a, b). We then co-transfected Flag-tagged STAT3 and HA-tagged PRTN3 into HEK293T cells, immunoprecipitated the cell lysates with anti-Flag or anti-HA
antibodies and immunoblotted the Co-IP complex. Our results indicated that STAT3 co-immunoprecipitated with PRTN3 but not with IgG in HEK293T cells (Fig. 2c, d). Furthermore, we investigated
whether PRTN3 could bind to other STAT family proteins by conducting experiments in which we overexpressed PRTN3 in HEK293T cells and tagged it with HA. We then immunoprecipitated the cell
lysates using anti-STAT1, anti-STAT2, and anti-STAT5 antibodies. Interestingly, we observed that PRTN3 was unable to bind to STAT1, STAT2, and STAT5 (Fig. s10a). We also investigated whether
neutrophil elastase (NE) and cathepsin G (CatG), which are active forms of neutrophil serine proteinases like PRNT3, could interact with STAT3. Specifically, we added STAT3 antibody into
HL60 cell lysates for immunoprecipitation with anti-NE and anti-CatG antibodies. The results indicated that there was no binding between STAT3 and NE or CatG (Fig. s10b). Moreover, the
results of the surface plasmon resonance (SPR) measurement showed that PRTN3 and STAT3 had a direct binding interaction in vitro, with a KD value of 2.24 × 10−7 M (Fig. 2e). We then
confirmed these findings in primary cells by conducting Co-immunoprecipitation (Co-IP) experiments on human primary CD34+ cells, which were isolated from cord blood using CD34-labeled beads.
Our results demonstrated that endogenous PRTN3 also directly interacted with STAT3 in primary cells (Fig. 2f, g). Additionally, confocal immunofluorescence analysis revealed that PRTN3 and
STAT3 were co-localized in the cytoplasm of human CD34+ cells (Fig. 2h). These findings strongly suggest that PRTN3 interacts with STAT3 and could potentially play a role in regulating
STAT3’s fate. PRTN3 BINDS TO THE N-TERMINAL DOMAIN OF STAT3 TO IDENTIFY THE CLEAVAGE SITE Based on the structural features that have been previously reported, STAT3 is composed of several
domains, including an N-terminal domain, linker domain, coiled-coil domain, DNA-binding domain, Src Homology 2 domain (SH2), C-terminal domain and transactivation domain [15, 16]. To
investigate the domains of STAT3 responsible for interacting with PRTN3, we generated Flag-tagged STAT3 mutants and transfected them into HEK293T cells (Fig. 3a). Subsequently, we performed
IP with an anti-Flag antibody and immunoblotting with an anti-HA antibody. Our analysis showed that the N-terminal domain of STAT3 played a significant role in its interaction with PRTN3
(Fig. 3c). Next, we generated an HA-tagged PRTN3 plasmid that included the full-length, N-terminal, and C-terminal domains (Fig. 3b), then performed a Co-IP experiment with Flag-tagged
STAT3, which validated our findings (Fig. 3d). In addition, we conducted an analysis to confirm the KD value of STAT3 with PRTN3 using recombinant human STAT3 with and without the N-terminal
domain. The results from surface plasmon resonance (SPR) indicated that the N-terminal domain of STAT3 had a direct binding affinity to PRTN3 (KD = 6.91 × 10−6 M), in contrast to other
domains of STAT3 (Fig. 3e). Next, we used the pure recombinant human STAT3 and PRNT3 protein for co-incubation to test whether PRTN3 could degrade STAT3 in vitro. After incubating STAT3 (1
μM) with hPRTN3 (1 μM), we observed the processing of the ~120 kDa monomer into two major fragments: ~90 kDa, as revealed by SDS-PAGE and silver staining (Fig. 3f). We predicted the presence
of PRTN3 cleavage sites in the N-terminal region segment of STAT3 and identified at least twenty-seven potential cleavage sites, denoted by an asterisk. Among these sites, we found that Val
(total 3) and Ala (total 13) were the preferred P1 residues for PRTN3 (Fig. 3g, Fig. s11a, b). The cleavage products were identified through mass spectrometry. The results revealed
variations in the cleavage site, and a hypersensitive site (Ala102 ↓-Arg103 ↓-Ile104 ↓-Val105 -Ala106 ↓-Arg107) was found in the N-terminal domain of STAT3 (Fig. 3f). Therefore, these
results indicate that PRTN3 binds directly to the N-terminal domain of STAT3 and cleaves the hypersensitive region. PRTN3 PROMOTES THE DEGRADATION OF STAT3-DEPENDENT OF UBIQUITINATION We
examined the levels of _Stat3_ mRNA in cells with overexpression or lack of PRTN3 and did not find any significant differences in _Stat3_ mRNA levels between the two groups (Fig. S12). Thus,
we hypothesized that PRTN3 might regulate the protein stability of STAT3 and conducted HEK293T cells were transfected with either PRTN3-HA tag or an empty vector for 24 h. Subsequently, the
cells were treated with the protein synthesis inhibitor cycloheximide (CHX) for a time-dependent period. The results of CHX chase assay showed that endogenous STAT3 in cells overexpressing
PRTN3 had a significantly shorter half-life (Fig. 4a). We also observed a significant increase in the half-life of endogenous STAT3 in down-regulated PRTN3 of human CD34+ cells following
siRNA treatment (Fig. 4b), indicating that PRTN3 plays a crucial role in decreasing STAT3 stability in vitro. To determine whether PRTN3 plays a role in regulating STAT3 stability through
inhibition of proteasome degradation, we found that overexpression of PRTN3 through PRTN3-HA tag transfection in HEK293T cells resulted in a reduction in the protein level of STAT3. This
reduction was subsequently recovered by treatment with proteasome inhibitor MG-132 (Fig. 4c). Conversely, depletion of PRTN3 in human CD34+ cells resulted in upregulation of STAT3 protein,
which was rescued by the addition of MG132 (Fig. 4d). These results suggest that PRTN3 plays a role in regulating STAT3 stability through the inhibition of proteasome degradation. We then
investigated whether PRTN3 affected the stability of STAT3 through ubiquitination and observed a significant decrease in the ubiquitin-mediated degradation of STAT3 in HEK293T cells that
overexpressed the PRTN3-HA tag (Fig. 4e). Conversely, downregulation of PRTN3 in human CD34+ cells resulted in an Increase in the levels of STAT3 protein ubiquitination (Fig. 4e).
Collectively, these results strongly suggest that PRTN3 plays a critical role in stabilizing STAT3. PRTN3 NEGATIVELY REGULATES STAT3-DEPENDENT MYELOID DIFFERENTIATION Previous studies have
demonstrated that G-CSF induces the activation of STAT3, which promotes myeloid differentiation both in vitro and in vivo [1, 17]. To investigate the physiological role of PRTN3 in
STAT3-dependent process. We treated HEK293T cells that overexpressed either wild type or PRTN3 with 10nM G-CSF for 24 h. Immunoblotting assay indicated that PRTN3 upregulation led to a
significant decrease in both total STAT3 and phosphorylated STAT3 expression in HEK293T cells treated with G-CSF, compared to WT cells (Fig. 5a). However, to further explore the involvement
of PRTN3 in G-CSF induced STAT3-regulated myeloid differentiation, we utilized primary c-Kit+ cells isolated from both _Prtn3__−/−_ and WT mice, following a 24-hour treatment with G-CSF. We
observed an increase in both STAT3 and P-STAT3 expression in _Prtn3__−/−_ primary c-Kit+ cells, compared to their WT counterparts (Fig. 5b). We then conducted immunofluorescence staining on
primary c-Kit+ cells treated with G-CSF for 2 h from both _Prtn3__−/−_ and WT mice to examine the nuclear translocation of STAT3, the results revealed a significant increase in the nuclear
translocation of STAT3 in the primary _Prtn3__−/−_ c-Kit+ cells compared to the WT cells (Fig. 5c). Next, we stimulated isolated primary c-Kit+ cells from both _Prtn3__−/−_ and WT mice with
G-CSF for 24 h to analyze downstream gene expression related to STAT3-dependent myeloid differentiation, specifically examining _P27__kpi1_ and _C/Ebpα_. The results showed that _Prtn3__−/−_
c-Kit+ cells treated with G-CSF exhibited higher levels of expression of _P27__kpi1_ and _C/Ebpα_ compared to WT c-Kit+ cells (Fig. 5d). To investigate the role of PRTN3 in the
G-CSF/STAT3-dependent mechanism during granulopoiesis and monopoiesis, we differentiated progenitor LSK cells (Lin-, Sca-1+, cKit+ cells) into myeloid cells using SCF and G-CSF. Within 3–4
days of cytokine stimulation, both granulocytic (CD11b + Ly6G+) and monocytic (CD11b + Ly6C+) cell populations developed successfully in this specific in this vitro cell culture system.
(Fig. S13a, b). The sorted stem/progenitor LSK cells from both _Prtn3__−/−_ and WT mice were cultured in an ex vivo myeloid culture system with the addition of SCF and G-CSF at different
time points. The results demonstrated a significant increase in the proportion of granulocytic as well as monocytic cell types when comparing Prtn3 knockout samples to those derived from WT
LSK counterparts within this particular myeloid culture setup. (Fig. 5e, Fig. S13c). These findings suggest that the absence of PRTN3 enhances myeloid differentiation by activating the
STAT3-dependent signaling pathway. STAT3 INHIBITOR RESCUES PRTN3-MEDIATED MYELOID DIFFERENTIATION To verify the involvement of the STAT3 pathway in the _Prtn3_-deficiency-induced myeloid
differentiation, we performed a pharmacological study using Stattic, a potent inhibitor of STAT3, both in vitro and in vivo. Specifically, we treated _Prtn3_-deficient LSK cells with the
small-molecule STAT3 inhibitor, which resulted in a significant reduction in STAT3 phosphorylation in both WT and _Prtn3_-deficient cells (Fig. 6a). The immunofluorescence staining of STAT3
and PRTN3 indicated that the upregulation of STAT3 nuclear translocation was reversed in the PRTN3-deficient LSK cells in comparison to the WT LSK cells (Fig. 6b, c). This observation was
further confirmed by analyzing the downstream gene expression of STAT3 in the LSK cells through qPCR, which included _C/EBPα_, _p27__kpi1_, _Bcl-X__L_, and _C-myc_ (Fig. 6d). To further
verify the role of STAT3 in PRTN3-mediated myeloid differentiation, we used flow cytometry to analyze the proportion of neutrophils and monocytes, which revealed a significant increase in
both neutrophils and monocytes in _Prtn3__−/−_ LSK cells compared to WT LSK cells. Furthermore, we found that Stattic effectively reversed the myeloid differentiation induced by _Prtn3_
depletion in LSK cells (Fig. 6e). We then administered Stattic (2 mg/kg) or a vehicle control orally via gavage three times per week for four weeks and analyzed the percentage of myeloid
cells every two weeks to assess the potential rescue effects of Stattic on myeloid differentiation in mice with Prtn3 deficiency (Fig. 6f). Our results showed that administering Stattic
every two weeks significantly ameliorated the elevated levels of myeloid cells in the peripheral blood of _Prtn3__−/−_ mice (Fig. S14). The analysis of myeloid cells in bone marrow using
flow cytometry indicated that the accumulation of myeloid cells induced by _Prtn3_ deficiency was significantly reduced by Stattic treatment (Fig. 6g). In conclusion, these findings provide
compelling evidence that Stattic effectively mitigates myeloid differentiation resulting from _Prtn3_ deficiency. PRTN3 DEFICIENCY RESTARTS THE MATURATION AND DIFFERENTIATION OF LEUKEMIC
BLAST To translate our findings into a clinically relevant setting, we evaluated the potential therapeutic benefits of reducing PRTN3 levels in acute myeloid leukemia. We extracted LSK cells
from both WT and _Prtn3__−/−_ mice and induced acute myeloid leukemia using MLL-AF9, then injected 1 × 105 AML cells into lethally irradiated hosts and monitored their survival rates to
determine the roles of _Prtn3_ deficiency in AML progression (Fig. 7a). The administration of _Prtn3_-knockout AML significantly extended survival up to five months in mice (Fig. 7b). To
further investigate the efficacy of this treatment, we examined leukemia cells in the peripheral blood of recipient mice four weeks after injection, showing a significant reduction in the
percentage of green fluorescent protein-positive (GFP+) AML cells in _Prtn3_-knockout AML mice (Fig. 7c, Fig. S15a). The spleens and livers of _Prtn_3-knockout AML mice were notably smaller
than those of control recipients (Fig. S15b, c). This observation was further supported by immunofluorescent staining (Fig. 7d, Fig. S15d), which indicated reduced infiltration in these
organs. The bone marrow of recipients with _Prtn3_-knockout AML was visibly redder than that of control mice (Fig. 7e). The recipients with _Prtn3_-knockout AML exhibited an increase in the
overall count of neutrophils and a decrease in the total number of monocytes, compared to the control recipients (Fig. S15e). Notably, the number of neutrophils with GFP+ was significantly
higher in the recipients with _Prtn3_-knockout AML than those with WT AML (Fig. 7f, Fig. S15f). To investigate whether the _Prtn3_-knockout induces the differentiation of neutrophils with
biology function, we evaluated the ability of phagocytosis and chemotaxis for neutrophil-like cells. The results showed that GFP+ neutrophils are capable of eliminating pathogens and can
migrate to the abdominal cavity via chemotaxis (Fig. 7g, h, Fig. S15g, h), indicating that AML cells with _Prtn3_ deficiency may differentiate into mature functional neutrophils. Next, we
examined the mRNA expression of _Prtn3_ in the AML cells of patients and found a significantly increased mRNA level of _Prtn3_ in the AML cells of patients compared with healthy controls
(Fig. 7i). We conducted further evaluations to determine whether depletion of _Prtn3_ enhances myeloid differentiation in human AML cell lines. We reduced the level of PRTN3 in HL60 cells
using siRNA, which was confirmed by immunoblotting and qPCR (Fig. 7j, k). Our findings indicate that the STAT3 protein increased in the cells treated with Si-_Prtn3_, and there was an
increase in the nuclear translocation of STAT3 in _Prtn3_-deficient cells (Fig. S16a). Moreover, the mRNA expression of downstream genes of STAT3, such as _C/Ebpɑ_ and _p27__kip1_, was
elevated in _Prtn3_-deficient cells compared to control cells (Fig. S16b). We analyzed differentiated myeloid cells using flow cytometry and found a significant increase in CD11b+ cells in
_Prtn3_-deficient cells compared to control cells, while CD14+ and CD15+ cells remained relatively unchanged (Fig. 7l, Fig. S16c). Additionally, the loss of _Prtn3_ cells showed an increase
in granulocyte-like cells, as observed through Wright-Giemsa staining (Fig. S16d). In NB4 cells, treatment with _Prtn3_-siRNA led to a decrease in PRTN3 protein levels, resulting in higher
nuclear translocation and upregulation of downstream genes, as confirmed by immunoblotting, immunofluorescence staining, and qPCR (Fig. S17 a–c). However, we observed that the loss of
_Prtn3_ in NB4 cells did not result in the presence of CD11b+, CD14+, CD15+ cells, or granulocyte-like cells compared to control NB4 cells (Fig. S17 d, e), as detected by flow cytometry and
Wright-Giemsa staining. Our findings indicate that inducing myeloid differentiation by decreasing PRTN3 is critical for AML-M2 rather than AML-M3. Furthermore, data mining on the publicly
available website Bloodspot (https://servers.binf.ku.dk/bloodspot) revealed that _Prtn3_ expression was dramatically elevated in most types of AML cells compared to healthy bone marrow (Fig.
S17 f). We analyzed the correlation between _Prtn3_ expression and overall survival in AML patients, as well as in different AML subtypes, utilizing the TCGA dataset
(https://portal.gdc.cancer.gov/projects) and the Kaplan-Meier Plotter (https://kmplot.com/analysis/index.php?p=service). The findings indicate a significant difference in _Prtn3_ expression
among AML patients based on FAB classification (_P_ < 0.05). Notably, AML patients with low _Prtn3_ expression and an intermediate karyotype demonstrated a higher survival probability
across various subtypes, including M1, M2, M3, and M5 (_P_ < 0.05) (Fig. S18). The results of our in vivo and in vitro studies, conducted on both mice and humans, suggest that targeting
PRTN3 could be a promising therapeutic approach for treating AML in a clinical setting. DISCUSSION In this study, we have demonstrated the role of PRTN3 in the regulation of myeloid
differentiation. Mechanistically, PRTN3 directly interacts with the N-terminal domain of STAT3, identifying cleavage sites, and thereby regulating STAT3 degradation through ubiquitination.
Consequently, it inhibits the expression of STAT3-dependent genes (Fig. 8). Altogether, this study reveals a novel role for PRTN3 in regulating myeloid differentiation and suggests its
potential as a target for treating acute myeloid leukemia. Numerous regulators of STAT3 have been well-established, including cytokine receptors [18], receptor tyrosine kinases [19],
G-protein-coupled receptors [20, 21], toll-like receptors [21,22,23], tyrosine phosphatases [24, 25], and the SOCS protein family [26, 27]. Our findings indicate that alterations in PRTN3
expression do not affect _Stat3_ mRNA levels. Instead, PRTN3 exerts post-transcriptional control by regulating STAT3 degradation, suggesting its role as a novel regulator of STAT3 at the
post-transcriptional level. Furthermore, the previous association between the PRTN3 protein and STAT3 is substantiated by our data [28], confirming the direct interaction between PRTN3 and
STAT3. Mass spectrum and CO-IP analyses demonstrate the specificity of this interaction with STAT3, distinguishing it from STAT1, STAT2, and STAT5,indicating that STAT3 is a specific target
of PRTN3 in signal transducer and activator of the transcription family. The STAT3 signal plays a central role in inducing myeloid differentiation in response to growth factors
[29,30,31,32]. Our findings provide substantiation for the role of PRTN3 in identifying cleavage sites on STAT3, resulting in ubiquitin-mediated degradation and subsequent reduction in
STAT3-dependent gene expression. This connection between PRTN3 and myeloid differentiation is particularly evident in G-CSF-stimulated LSK cells. Our data suggest that PRTN3 is a viable
candidate for targeted regulation of myeloid differentiation, potentially avoiding side effects associated with direct manipulation of STAT3. Neutrophil serine proteases, such as neutrophil
elastase (NE), cathepsin G (CG), and PRTN3, have been implicated in the regulation of inflammatory conditions [33]. Our investigation reveals that PRTN3 displays specificity for STAT3
compared to NE and CG, suggesting the need for further studies regarding the functions of NE and CG within the signal transducer and activator of the transcription family. Furthermore, PRTN3
is expressed in hematopoietic processes, encompassing hematopoietic stem and progenitor cells [9] as well as mature myeloid cells [8, 10, 34, 35]. Previous reports have indicated that PRTN3
regulates neutrophil apoptosis during inflammatory responses through the targeting of pro-caspase-3 [8] and modulates the HSPC population through apoptosis [9]. Our findings suggested that
further studies are warranted to investigate the role of PRTN3 in diverse hematopoietic cell types, which will enhance our understanding of protease functions. It has been reported that the
downregulation of PRTN3 expression in response to treatment with All-trans retinoic acid (ATRA) plus arsenic trioxide (ATO), leads to the differentiation of leukemia cells capable of ROS
production and phagocytosis [36, 37]. While previous studies did not investigate the underlying mechanism [36], our findings suggest that PRTN3 regulates STAT3-dependent myeloid
differentiation in the context of acute myeloid leukemia (AML). STAT3 serves as a pivotal molecule in anti-AML immunity, particularly through T cells. Our findings unequivocally show that
the depletion of PRTN3 has no impact on T cell functions, encompassing proliferation, activation, and anti-AML immunity, strongly suggesting that PRTN3 controls AML differentiation rather
than influences T cell immunity in AML. The extent of differentiation varies across AML subtypes, with AML M2 displaying the most notable response to PRTN3 deregulation in cell lines.
Intriguingly, AML patients with low _Prtn3_ expression exhibit a higher probability of survival across various subtypes, including M1, M2, M3, and M5, indicating that inhibiting PRTN3 could
potentially lead to a substantial improvement in the clinical survival of AML patients. Significantly, our data also reveal that targeting PRTN3 inhibition promotes the differentiation of
AML cells into neutrophils capable of phagocytosis and chemotaxis migration, hinders malignant proliferation, induces cellular maturation, significantly enhances survival rates, and reduces
AML cell counts in mice with MLL-AF9-induced AML, underscoring PRTN3 as a promising therapeutic target for AML. However, although targeting PRTN3 holds promise as a novel therapeutic
approach for AML, direct targeting with small molecules faces challenges. Thus, further investigations, encompassing both fundamental and clinical studies, are warranted to evaluate the
therapeutic potential of inhibiting PRTN3 for the treatment of AML. MATERIALS AND METHODS MICE _Prtn3__−/−_ mice were generated by GemPharmatech Inc (Jiangsu, China) using CRISPR/Cas9
technology, as described previously [38]. Tails were got from offspring, and the primers used for _Prtn3_ genotyping were forward: 5′- CCCTGATCCACCCGAGATTC-3′ and reverse 5′-
GGTTCTCCTCGGGGTTGTAA -3′. _Prtn3__flo/flox_ mice (Strain #:030761) were obtained from Jackson Laboratories. LysMcre mice (Strain #:004781) were purchased from Jackson Laboratories. The
myeloid lineage conditional _Prtn3_ knockout mice (_Prtn3__LyzKO_) were crossed by LysMcre mice and _Prtn3__flo/flox_ mice. The primers used for _Prtn3__flox/+_ genotyping were forward: 5′-
GGT CTG AAC TGA CAG CAA AGC-3′ and reverse 5′- CCC TAA CCA CTC CCC TAT CC-3′. the primers used for LysMcre genotyping were common: 5′- AAG GAG GGA CTT GGA GGA TG -3′, wild type reverse 5′-
GTC ACT CAC TGC TCC CCT GT -3′, Mutant Reverse 5′- ACC GGT AAT GCA GGC AAA T -3′. Six-to-eight-week-old C57BL/6 CD45.2 mice and CD45.1 mice were purchased from Beijing Vital River
Laboratories. In all experiments with knockout mice, corresponding littermates as WT controls were used. All animals were maintained at the Animal Core Facility of the State Key Laboratory
of Experimental Hematology. THE MOUSE MODEL OF HEMATOPOIETIC STEM CELL TRANSPLANTATION Mice carrying CD45.1 (Recipient) were irradiated with single doses of 9.5 Gy. For hematopoietic stem
cell total transplantation, recipients were injected with 1 × 103 sorted LSK cells from the donor (CD45.2) and 5 × 105 BM cells from the donor (CD45.1). Transplanted into recipient mice
after myelopoiesis by tail vein injection. Mice are given oral enrofloxacin for 4 weeks after transplantation. Mice are Sacrificed 4 months after transplantation, the femur, tibia, ilium,
and spleen were harvested, and bone marrow cells are flushed out with PBE by 1 mL syringe, filtered, and resuspended. Lysis of red blood cells of the bone marrow with erythrocyte lysate, the
chimerism was analyzed at the end of each experiment. Flow cytometry antibody [CD45.1-PE (Biolegend, 110708), CD45.2-APC (Biolegend, 109814)] labeling is performed and finally analyzed by
flow cytometry. At the same time, bone marrow and liver frozen sections with a thickness of 7 μm were prepared, corresponding immunofluorescence staining was performed, and the ratio of
CD45.1 to CD45.2 was observed by confocal microscope. GENERATED AML MOUSE MODEL For transplantation of _Prtn3__−/−_ leukemia cells, 2 × 105 MLL-AF9 transduced lineage cells from WT or
_Prtn3__−/−_ mice were injected intravenously into half-lethally irradiated (4.5 Gy) C57BL/6 recipient mice. Then, WT or _Prtn3__−/−_ AML cells were further sorted from primary recipient
mice, and then, the cells were transplanted with 2 × 105 BM into half-lethally irradiated (4.5 Gy) C57BL/6 recipient mice. STAT3 INHIBITOR MOUSE MODEL Mice aged at 8 weeks were randomized to
receive either Stattic (2 mg/kg, p.o. #T6308, TargetMol, USA) or vehicle alone (5%DMSO, 95% peanut oil) for 4 weeks. 6 mice in each group were sufficient for our experiments. MOUSE MODEL OF
_E. COLI_-INDUCED PERITONITIS FOR TESTING CHEMOTAXIS MIGRATION E. coli cultures were grown in Luria-Bertani (LB) liquid media overnight and reached the exponential growth phase (0.5 OD600)
[39]. Then, washed three times with sterile normal saline. Then AMLPrtn3KO mice were injected intraperitoneally (i.p.) with 1 × 106 _E. coli_ in a total volume of 500 μl saline. Six hours
after bacterial infection, mice were anesthetized. Peritoneal lavage was performed using 10 ml of normal PBS that contained 0.1% bovine serum albumin. The cells were incubated with
antibodies and detected by flow cytometry. CELLS HEK293T was cultured in DMEM (ThermoFisher) supplemented with 10% (v/v) FBS (Hyclone, UK) at 37 °C. HEK293T cell lines were purchased from
ATCC (Manassas, USA). HL-60 cells were cultured in IMDM medium (Gibco) supplemented with 2% (v/v) FBS (Hyclone, UK) at 37 °C. NB4 cells were cultured in 1640 medium (ThermoFisher)
supplemented with 10% FBS (Hyclone, UK) at 37 °C. HL-60 cell lines and NB4 cell lines were gifted from Dr. Guoguang Zheng (Tianjin, China). All cells were grown at 37 °C in a 5% CO2
incubator (Thermo Fisher, USA). Cells were transfected with Lipofectamine RNAiMAX (Invitrogen, USA) was used for the transfection of plasmids or siRNAs into cells. PLASMID EXAMINATION, CELL
CULTURE, VIRUS PRODUCTION, AND TRANSDUCTION An MSCV-MLL-AF9-IRES-GFP plasmid was gifted from Dr. Tao Cheng and Dr. Weiping Yuan at the State Key Laboratory of Experimental Hematology,
National Clinical Research Center for Blood Diseases [40]. We re-clone the MLL fragment by qPCR (Fig S 17a). The mRNA was isolated from AMLPrnt3KO mice to determine two key AF9 genes,
_Hoxa9_ and _Meis1_, to confirm the plasmid by qPCR (Fig S 17b). The primer as follows: ALL-AF9 F: AAC CAC CTC CGG TCA ATA AGC; R: TTC ACG ATC TGC TGC AGA ATG; Hoxa9 F: GGA ATA GGA GGA AAA
AAC AGA AGA GG; R: TGT ATG AAC CGC TCT GGT ATC CTT; Meis1: F: TCA CCA CGT TGA CAA CCT CG; R: GCT TTC TGC CAC TCC AGC TG. An MSCV-MLL-AF9-IRES-GFP plasmid together with pKat and pVSVG
packaging plasmids was transfected into 293T cells using Lipofectamine 3000 (ThermoFisher) to transform normal hematopoietic stem and progenitor cells into AML cells [41]. After 72 h of
culture, the supernatant containing retroviruses was collected and concentrated BY an Amicon filter (Millipore). normal hematopoietic stem and progenitor cells from WT and _Prtn3__−/−_ mice
were enriched by lineage cell depletion beads (Miltenyi), and then transduced with MLL-AF9 retroviruses in the presence of 4 μg/ml polybrene (Sigma). The cells were incubated in IMDM (Gibco)
with 15% FBS, 50 ng/ml mouse SCF (Peprotech), 10 ng/ml mouse IL-3 (Peprotech), and mouse IL-6 (Peprotech) for 2 days. Prepare whole bone marrow single-cell suspension of experimental mice
(donor, CD45.2), perform positive enrichment of c-kit magnetic beads, and sort hematopoietic stem cell LSK (Lin-c-Kit + Scal1+) cells. Sacrificed CD45.1 mouse and prepare a whole bone marrow
single-cell suspension. ISOLATED PRIMARY HUMAN CD34+ HPCS Primary CD34+ hematopoietic progenitor cells (HPCs) were isolated from the blood of the human umbilical cord obtained from Blood
Diseases Hospital. In brief, using magnetic bead separation and viably frozen as previously described [42], red blood cells first were removed from total blood, and then hematopoietic cells
were purified by Ficoll gradient centrifugation. CD34+ HPCs were purified using a Dynabeads™ CD34 progenitor cell selection system (Invitrogen™, 11301D), according to the manufacturer’s
instructions. Frozen cells were thawed and recovered overnight in stem cell media or prepared for protein extraction. ISOLATED PRIMARY HUMAN AML CELLS The human AML cell samples were
obtained from Blood Diseases Hospital. In brief, using magnetic bead separation and viably frozen as previously described [42], red blood cells were removed from total blood, and then
hematopoietic cells were purified by Ficoll gradient centrifugation. BONE MARROW CULTURE Bone marrow cells were harvested from the tibia, femur, and pelvic bones by crushing the bones with a
mortar and pestle in PBE [43]. Cells gathered were filtered through a 70 μm filter to make a single cell suspension and were enriched for progenitor cells using mouse hematopoietic
progenitor cell isolation kit (BioLegend, San Diego, CA, USA) and cultured in DMEM media with 10% FBS. Cell culture media were supplemented with stem cell factor (SCF: 50 ng/ml); FLT3L (10
ng/ml); and thrombopoietin (TPO: 10 ng/ml) (BioLegend, San Diego, CA, USA). LSK EXPANSION CULTURE Sorted LSK cells were cultured at a concentration of 104 cells per 1 mL media supplemented
with SCF (50 ng/ml) and TPO (50 ng/ml) in a single well of a 12-well tissue culture plate. The choice and concentration of cytokines were based on the stem cell expansion protocol as
previously reported [43]. LSK expansion was a culture in Serum-free StemSpan SFEMII medium (STEMCELL Technologies, Vancouver, Canada), with flow analysis on the second, third, or fourth day
post-LSK culture. MYELOID CULTURE Sorted LSK cells were cultured in DMEM media supplemented with SCF (50 ng/ml) and G-CSF (10 ng/ml) in a 48-well tissue culture plate. The choice and
concentration of cytokines were based on the myeloid differentiation protocol as described [44]. DMEM media supplemented with 10% FBS was used for cell culture media. Cells were analyzed
after the culture of LSK cells by flow cytometry. CFSE T CELL PROLIFERATION ASSAY For the T cell proliferation assay, anti-CD3-preactivated T cells were first labeled with CFSE (Invitrogen;
Cat No. C34554) in a dilution of 1:1000. After 3 days of co-culture, T cells were harvested and CFSE density was measured by flow cytometry. Each experiment was performed in triplicate. T
CELL ACTIVATION BIOASSAY For T cell activation, the T cells from the spleen were labeled by CD62I (Biolegend, 104405), CD69 (Biolegend, 164203), IFN-γ (Biolegend, 505807), and TNF-α
(Biolegend, 506305) for 30 min on ice, then measured by flow cytometry. T-CELL KILLING ASSAY Mice spleen T cells were isolated and cultured in RPMI-1640 medium and activated with Dynabeads™
Mice T-Activator CD3/CD28 (Gibco, CA, USA) and 10 ng/ml IL-2 for 3 days according to the manufacturer’s instructions. AF9-AML cells were seeded into 12-well plates at a cell-dependent
concentration. After 24 h, activated T cells were cocultured with AF9-AML cells for 24 h at a ratio of 10:1. Cell debris was removed, and cells were harvested and labeled with annexin V and
PI for fluorescence-activated cell sorting (FACS) analysis. SIRNA-MEDIATED GENE KNOCKDOWN human primary CD34+ cells, HL-60 cell line, and NB4 cells line were transfected with non-targeting
control siRNAs (sc-44230, Santa Cruz), or _Prtn3_ siRNA (sc-42968, Santa Cruz). 6 μL LipofectamineTM RNAiMAX (13778075, Thermo FisherScientific) was used as the transfection reagent based on
the manufacturer’s protocols. Cells were harvested 48 h after transfection. The efficiency of _Prtn3_ knockdown was analyzed by Western Blot. COMPLETE BLOOD COUNT The peripheral blood (50
μL) was collected by heparinized capillary tubes (Fisher Scientific) and transferred into K2-EDTA-coated tubes (Becton Dickinson). Blood parameters were analyzed by using Hemavet 950FS (Drew
Scientific). PHAGOCYTOSIS ASSAY Fluorescein conjugate E. coli (K-12 strain) BioParticles (Molecular Probes, E2861, PE-labeled heat-killed E. coli) were diluted in serum media and incubated
for 30 min, followed by two washes in PBS. Next, PE-labeled, heat-killed E. coli was added to opti-MEM medium with 10% FCS and incubated with GFP+ cells from AMLPrtn3KO mice at different
time points, 0, 0.5, 1, 3, 5 h in a humidified atmosphere of 5% CO2 at 37 °C, and GFP+ cells were washed twice with PBS, and then staining with Ly6G-APC antibody, followed by flow cytometric
analysis [45]. PLASMIDS Human _Stat3_ cDNA (RDC1454) was kindly obtained from Biotechne and cloned into the pcDNA3.1 vectors, respectively. Flag-tagged Stat3 (1-770), Flag-tagged Stat3
(1-688), Flag-tagged Stat3 (1-583), Flag-tagged Stat3 (1-465), Flag-tagged Stat3 (1-320), Flag-tagged Stat3 (1-130) were subcloned from pcDNA3.1-STAT3-Flag as described previously [46].
N-terminal domain or C-terminal domain _Prtn3_ were amplified from the HA-tagged full-length _Prtn3_, which were then subcloned into the pcDNA3.1 vectors. All constructs were confirmed by
DNA sequencing. HA-Ubiquitin plasmid (Plasmid #18712) was purchased from the addgene. FLOW CYTOMETRY AND CELL SORTING BM cells from femurs and tibias were flushed into PBE supplemented with
2% FBS (Atlanta Biologicals, S11150H). Red blood cells were lysed by eBioscience™ 1X RBC Lysis Buffer (Invitrogen™, 00-4333-57). Ten million BM cells were incubated with an antibody mixture
including antibodies for lineage markers: Ly-6C-FTIC (Biolegend, 128005), Ly-6G-PE-Cy7 (Biolegend, 127617), CD3-PE (Biolegend, 980008), CD11b-APC (Biolegend, 101212), CD45R/B220- PE-Cy5
(Biolegend, 103209), as well as Sca1-PE/Cy7 (Biolegend, 108114) and c-Kit-APC/Cy7 (Biolegend, 105826) in DMEM (Life Technologies, 31053-028) supplemented with 2% FBS. For LSK subset
analysis, Lin-APC (Biolegend, 348703), Sca1-PE/Cy7 (Biolegend, 108114) and c-Kit-APC/Cy7 (Biolegend, 105826). CD34-FITC (eBioscience, 11-0341-85) and CD135-PE (Biolegend, 135306) were used.
For LK subsets, CD34-FITC and CD16/32-PE (Biolegend, 101308) were used. Samples were incubated on ice for 30 min, then washed and filtered before analysis. Samples were stained for one hour
on ice for CD34 staining. Data were collected on FACSCanto II or LSR II flow cytometers (Becton Dickinson) and analyzed by FlowJo software (Tree Star). For sorting highly purified cells, an
immunomagnetic negative selection kit was used to enrich HSPCs (Stem Cell Technologies, 19756). Enriched cells were stained with Lin-APC (Biolegend, 348703), c-Kit-PE (Biolegend, 105808),
and Sca1- PE/Cy7 (Biolegend, 108114). DAPI (BD Biosciences, 564907) was added to exclude dead cells. Cells were sorted on an Aria (Becton Dickinson) or MoFlo (Dako) cell sorter. PROTEIN
PREPARATION, IMMUNOPRECIPITATION, AND IMMUNOBLOTTING ANALYSIS For immunoprecipitation and ubiquitination analysis, the total cellular protein was isolated with a cell lysis buffer (Cell
Signaling Technology; #9803). Cell lysates were incubated with the appropriate primary antibody [anti-HA (Invitrogen, 26183), anti-FLAG (Invitrogen; 26183), anti-STAT3(Cell Signaling
Technology, 9139), anti-PRTN3 (Abcam, ab103632)] overnight at 4 °C followed by 1.5 h incubation with protein A/G agarose beads. The beads were washed three times with the lysis buffer and
were eluted in 5 × SDS/PAGE loading buffer for immunoblotting. For immunoblotting analysis, cells were lysed with cell lysis buffer supplemented with protein inhibitors (Roche, Switzerland).
The cell lysates were incubated with the following antibodies: anti-HA (Invitrogen, 26183), anti-FLAG (Invitrogen; 26183), anti-STAT1(Cell Signaling Technology, 9172), anti-STAT2 (Cell
Signaling Technology, 4597), anti-STAT3 (Cell Signaling Technology, 9139), anti-STAT5 (Cell Signaling Technology, 94205), anti-PRTN3 (Abcam, ab103632), anti-Phospho-STAT3 (Cell Signaling
Technology, 9145), anti-Neutrophil Elastase Antibody (Cell Signaling Technology, 63610), anti-Cathepsin G (Abcam, ab192793) and anti-β-Actin (Cell Signaling Technology, 4967), overnight at 4
°C. After washing with TBST three times, the membranes were incubated with second antibody (Goat Anti-Mouse IgG (H + L)-HRP Conjugate; 1706516; Bio-Rad; Goat Anti-Mouse IgG (H + L)-HRP
Conjugate, 1706516; Bio-Rad) (dilution at 1:10,000) at room temperature for 90 min. Images were visualized using an ChemiDoc (Bio-Rad). For quantification of the Immunoblotting assay,
densitometric analysis of bands of target proteins and loading controls (β-Actin) was performed with NIH Image J software as we described previously [47]. Western blotting was performed in
three independent experiments. RECOMBINANT PROTEIN PREPARATION The STAT3 recombinant protein (Ab43618) was ordered from Abcam. The STAT3 recombinant protein (ST3-H5149-100 μg) was ordered
from ACRO. The STAT3 recombinant protein (HY-P70574) was ordered from ACE. The PRTN3 recombinant protein (HY-P70509) was ordered from ACE. All proteins were prepared for surface plasmon
resonance (SPR) measurement. IMMUNOFLUORESCENCE (IF) STAINING AND WRIGHT-GIEMSA STAINING Formaldehyde-fixed, paraffin-embedded liver and bone samples were sectioned into 7 μm slides.
immunofluorescence (IF) staining was performed as previously described [48]. Briefly, formaldehyde-fixed, OCT-embedded slides for tissue samples were subjected to antigen retrieval, followed
by blocking and antibody (anti-STAT3 (Cell Signaling Technology, 9139), and anti-PRTN3 (Abcam, ab103632)) (1:100) incubation overnight. For in vitro experiments, cells were seeded on
coverslips and were fixed with 4% paraformaldehyde, permeated with 0.1% Triton X- 100, and then blocked with 10% BSA. Primary antibodies (anti-STAT3 (Cell Signaling Technology, 9139), and
anti-PRTN3 (Abcam, ab103632)) (1:100) were diluted as suggested and incubated at 4 °C overnight in a moist chamber. The slides were then washed with PBS and incubated with 488-/546
-conjugated secondary antibodies. Slides were further incubated with DAPI (Invitrogen) and mounted in IF mounting medium (Service Bio). Quantification of the mean fluorescence intensity was
performed with the ImageJ software under at least three randomly selected fields. Confocal microscopy was performed using a Spinning disk confocal microscopy system (Perkinelmer, ultraVIEW
VOX). For Wright-Giemsa staining, Cells (2 × 105 cells/mL) were prepared. Then, cells were harvested and the density was adjusted to 2 × 104 cells/mL. The cells were collected and used
Wright–Giemsa (Leagene, Beijing, China) staining according to the manufacturer’s protocol on slides prepared by cytospin to perform a morphological assessment. The morphology of cells was
observed under a light microscope. COLONY-FORMING CELL ASSAYS Colony formation assays were performed as previously reported [9]. LSK cells (2 × 104) from WT and knockout mice were seeded in
semisolid Methocult GF M3434 medium containing SCF, IL-3, IL-6, and Epo for detection of colony-forming units-granulocyte, monocyte, and burst-forming units-erythroid (Stem Cell
Technologies, 03434). Colony numbers were counted on day 7, and images for colony sizes were obtained on day 8. ANALYSIS OF THE CORRELATION BETWEEN _PRTN3_ AND PATIENT SURVIVAL IN THE TCGA
DATABASE An online tool Kmplot (https://kmplot.com/analysis/index.php?p=service) was used to determine the correlation between _Prtn3_ and patient survival, as well as the _Prtn3_ level from
patients with M0-M7 was collected from the TCGA database (https://portal.gdc.cancer.gov/projects). SURFACE PLASMON RESONANCE (SPR) MEASUREMENT To measure the binding affinities of PRTN3
with the STAT3, The N-terminal domain of STAT3, and STAT3 without the N-terminal domain, an SPR-based Biacore K8 biosensor (Biacore AB, Uppsala, Sweden) with CM5 was used as the sensor chip
as previously reported [49]. The target proteins were diluted to a final concentration of 20 μg/mL in 10 mmol in PBS (pH 7.2) and then immobilized to CM5 by the standard primary amine
coupling method at 25 °C. Firstly, The CM5 surface was activated by injecting a 1:1 mixture containing 200 mmol/L 1-ethyl-3-(3-dimethylamino propyl) carbodiimide hydrochloride and 50 mmol/L
N-hydroxysuccinimide for a target level of 10,000 RU at a flow rate of 20 μL/min. The target proteins were injected into the CM5 surface. All the screening assays were performed over the
unmodified dextran surface and the protein surface. Each sample assay consisted of a 180 s buffer injection and a 300 s dissociation phase and the blank injection was used to check the
carryover effects. The signal was adjusted for nonspecific binding of the samples to the dextran matrix by subtracting the signal in the reference channel from the signal in the active
channel. The experimental data were fitted and analyzed using the BIA evaluation software (Cytiva, Sweden) by steady-state analysis. ELECTROPHORESIS A STAT3 (1 μM final) was incubated with
hPR3 (1 μM final) for 30–120 min at 37 °C in PBS solution (pH 7.2). The mixtures were then denatured/reduced by the addition of reducing buffer (19, b-mercaptoethanol) and then boiled for 5
min at 95 °C. The mixtures were separated on a 10% SDS-PAGE gel at room temperature for about 1 h and visualized by silver nitrate (Invitrogen, Carlsbad, CA, USA) staining according to the
manufacturer’s protocol. Size markers (25–170 kDa) were used (Thermo Fisher Scientific, Waltham, MA, USA). The bands representing the proteins according to their size appeared dark brown on
the gel. MOLECULAR DOCKING As previously reported [50], The in-silico analysis of the protein-ligand binding mode was performed with the crystal structure of hPR3 used as a receptor
(1FUJ.pdb) [51]. The models of ligand structure were prepared and optimized using ChemBio3D 12.0. The protonation and atom types of all molecules were set with SPORES. MASS SPECTROMETRY Mass
spectrometry was performed as previously described [52]. Briefly, harvested cell lysates were incubated with the anti-HA primary antibodies overnight at 4 °C, and conjugated with protein
A/G beads (Santa Cruz Biotechnology, CA) for 1.5 h. After washing, immunoprecipitants were boiled in Laemmli sample buffer for 5 min. The immunoprecipitated proteins were detected by reverse
phase liquid chromatography/mass spectrometry (RPLC/MS)-ESI-Q-ToFQ analyzer (TripleTOF 6600 MS system, Applied Biosystem, USA). MALDI-TOF MS STAT3 samples were analyzed on an UltraFlex I
mass spectrometer (Bruker Daltonics). A cysteine reduction was carried out for 10 min at 37 °C using 5 mM Tris(2-carboxyethyl) phosphine hydrochloride prior to MS analysis of whole STAT3
samples. Samples were diluted 50-fold or 100-fold in a solution of 4HCCA saturated in a solution of 66.6% water, 33.3% acetonitrile, and 0.1% trifluoroacetic acid. Matrix/sample solutions
were spotted onto a gold-plated sample probe using the ultrathin layer method [50]. MALDI-TOF- MS spectra were processed and annotated using FLEXANALY- SIS 3.3 (Bruker Daltonics) and PAWS
8.5 (ProteoMetrics) software, respectively. QUANTITATIVE RT-PCR Total RNA was prepared from neutrophils, LSK cells, HL60 and NB4 cells using TRIzol (Invitrogen). cDNA was generated using 1
μg total RNA using a High-Capacity cDNA Reverse Transcription Kit from Applied Biosystems. Quantitative polymerase chain reaction (qPCR) was performed on the CFX-96 real-time PCR detection
system (Bio-Rad, Hercules, CA, USA) with iQ SYBR Green Supermix (1708880B10, Bio-Rad) under the following conditions: 94 °C for 10 min; 40 cycles of 94 °C for 15 s, 58 °C for 30 s, 72 °C for
30 s; and final elongation at 72 °C for 15 min. Relative mRNA expression to that of the housekeeping gene β-Actin was calculated. Data were normalized and the control group was set at 1.0.
Gene expression was then measured with a CFX96 Real-Time PCR System (Bio-Rad) using the following primer pairs: Prtn3(Human): F 5ʹ- CCTGCAGGAGCTCAATGT-3ʹ; R 5ʹ- CTG AGT CTC CGA AGC AGA TG-3ʹ
; Prtn3(Ms): F 5ʹ- CTT GAT CTG CAA TGG CAT TCT T-3ʹ; R 5ʹ- GGC GAA GAA ATC AGG GAA CT-3ʹ ; Stat3 (Human): F 5ʹ- GAG AAG GAC ATC AGC GGT AAG-3ʹ; R 5ʹ- CAG TGG AGA CAC CAG GAT ATT G-3ʹ;
C/EBPɑ (Human): F 5ʹ- GAA GTC GGT GGA CAAG AAC A-3ʹ; R 5ʹ- TCA TTG TCA CTG GTC AGC TC -3ʹ; C/EBPɑ(Ms): F 5ʹ- CAA GAA GTC GGT GGA CAA GAA-3ʹ; R 5ʹ- CGT TGC GTT GTT TGG CTT TA-3ʹ ;
P27kpi1(Human): F 5ʹ- CTA ACTC TGA GGA CAC GCA TTT-3ʹ; R 5ʹ-TGC AGG TCG CTT CCT TAT TC-3ʹ; P27kpi1(Ms): F 5ʹ- AGC TTG CCC GAG TTC TAC TA -3ʹ; R 5ʹ- GAG TTT GCC TGA GAC CCA ATT A -3ʹ;
BcL-XL(Human): F 5ʹ- GGT GGT TGA CTT TCT CTC CTA C -3ʹ; R 5ʹ- TCT CCG ATT CAG TCC CTT CT-3ʹ; BcL-XL(Ms): F 5ʹ- TGG TCG ACT TTC TCT CCT ACA-3ʹ; R 5ʹ- CCC TCT CTG CTT CAG TTT CTT C-3ʹ; C-myc
(Human)F 5ʹ- CTG AGG AGG AAC AAG AAG ATG AG -3ʹ; R 5ʹ- TGT GAG GAG GTT TGC TGT G-3ʹ; C-myc(Ms) F 5ʹ- CTC CGT ACA GCC CTA TTT CAT C -3ʹ; R 5ʹ- TGG GAA GCA GCT CGA ATT T-3ʹ; β-Actin (Ms): F
5ʹ-AGC CAT GTA CGT AGC CAT CCA-3’; R 5ʹ- TCT CCG GAG TCC ATC ACA ATG-3ʹ; Gapdh (Human): F 5ʹ- AGG GCT GCT TTT AAC TCT GGT-3ʹ; R 5ʹ-CCC CAC TTG ATT TTG GAG GGA-3ʹ. STATISTICAL ANALYSIS Each
experiment was performed at least three times. All experiment data were analyzed using GraphPad Prism 9.0 (GraphPad Software Inc. USA) and were presented as the mean ± SD. Statistical
analysis was performed using Student’s _t_ test, one-way ANOVA, or two-way ANOVA. A value of _P_ < 0.05 was considered statistically significant. DATA AVAILABILITY Detailed information on
antibodies and reagents used in the manuscript is provided in the Materials and Methods. Raw western blots are available in the Supplemental file. REFERENCES * Miranda MB, Johnson DE.
Signal transduction pathways that contribute to myeloid differentiation. Leukemia. 2007;21:1363–77. Article CAS PubMed Google Scholar * Kovtonyuk LV, Caiado F, Garcia-Martin S, Manz EM,
Helbling P, Takizawa H, et al. IL-1 mediates microbiome-induced inflammaging of hematopoietic stem cells in mice. Blood. 2022;139:44–58. Article CAS PubMed Google Scholar * Boettcher S,
Manz MG. Regulation of inflammation- and infection-driven hematopoiesis. Trends Immunol. 2017;38:345–57. Article CAS PubMed Google Scholar * Ying M, Shao X, Jing H, Liu Y, Qi X, Cao J,
et al. Ubiquitin-dependent degradation of CDK2 drives the therapeutic differentiation of AML by targeting PRDX2. Blood. 2018;131:2698–711. Article CAS PubMed Google Scholar * Ngo S,
Oxley EP, Ghisi M, Garwood MM, McKenzie MD, Mitchell HL, et al. Acute myeloid leukemia maturation lineage influences residual disease and relapse following differentiation therapy. Nat
Commun. 2021;12:6546. Article CAS PubMed PubMed Central Google Scholar * Humbert M, Seiler K, Mosimann S, Rentsch V, Sharma K, Pandey AV, et al. Reducing FASN expression sensitizes
acute myeloid leukemia cells to differentiation therapy. Cell Death Differ. 2021;28:2465–81. Article CAS PubMed PubMed Central Google Scholar * Sykes DB, Kfoury YS, Mercier FE, Wawer
MJ, Law JM, Haynes MK, et al. Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell. 2016;167:171–186 e115. Article CAS PubMed
PubMed Central Google Scholar * Loison F, Zhu H, Karatepe K, Kasorn A, Liu P, Ye K, et al. Proteinase 3-dependent caspase-3 cleavage modulates neutrophil death and inflammation. J Clin
Investig. 2014;124:4445–58. Article PubMed PubMed Central Google Scholar * Karatepe K, Zhu H, Zhang X, Guo R, Kambara H, Loison F, et al. Proteinase 3 limits the number of hematopoietic
stem and progenitor cells in murine bone marrow. Stem Cell Rep. 2018;11:1092–105. Article CAS Google Scholar * Cheung P, Schaffert S, Chang SE, Dvorak M, Donato M, Macaubas C, et al.
Repression of CTSG, ELANE and PRTN3-mediated histone H3 proteolytic cleavage promotes monocyte-to-macrophage differentiation. Nat Immunol. 2021;22:711–22. Article CAS PubMed PubMed
Central Google Scholar * Akhtar J, Jain V, Kansal R, Priya R, Sakhuja P, Goyal S, et al. Quantitative tissue proteome profile reveals neutrophil degranulation and remodeling of
extracellular matrix proteins in early stage gallbladder cancer. Front Oncol. 2022;12:1046974. Article CAS PubMed Google Scholar * von Richthofen HJ, Westerlaken GHA, Gollnast D,
Besteman S, Delemarre EM, Rodenburg K, et al. Soluble signal inhibitory receptor on leukocytes-1 is released from activated neutrophils by proteinase 3 cleavage. J Immunol. 2023;210:389–97.
Article Google Scholar * Jerke U, Eulenberg-Gustavus C, Rousselle A, Nicklin P, Kreideweiss S, Grundl MA, et al. Targeting cathepsin C in PR3-ANCA vasculitis. J Am Soc Nephrol.
2022;33:936–47. Article CAS PubMed PubMed Central Google Scholar * Chu TY, Zheng-Gerard C, Huang KY, Chang YC, Chen YW, I KY. et al. GPR97 triggers inflammatory processes in human
neutrophils via a macromolecular complex upstream of PAR2 activation. Nat Commun. 2022;13:6385 Article CAS PubMed PubMed Central Google Scholar * Miklossy G, Hilliard TS, Turkson J.
Therapeutic modulators of STAT signalling for human diseases. Nat Rev Drug Discov. 2013;12:611–29. Article CAS PubMed PubMed Central Google Scholar * Brambilla L, Genini D, Laurini E,
Merulla J, Perez L, Fermeglia M, et al. Hitting the right spot: mechanism of action of OPB-31121, a novel and potent inhibitor of the Signal Transducer and Activator of Transcription 3
(STAT3). Mol Oncol. 2015;9:1194–206. Article CAS PubMed PubMed Central Google Scholar * McLemore ML, Grewal S, Liu F, Archambault A, Poursine-Laurent J, Haug J, et al. STAT-3 activation
is required for normal G-CSF-dependent proliferation and granulocytic differentiation. Immunity. 2001;14:193–204. Article CAS PubMed Google Scholar * Yuan J, Zhang F, Niu R. Multiple
regulation pathways and pivotal biological functions of STAT3 in cancer. Sci Rep. 2015;5:17663. Article CAS PubMed PubMed Central Google Scholar * Murone M, Vaslin Chessex A, Attinger
A, Ramachandra R, Shetty SJ, Daginakatte G, et al. Debio 0617B inhibits growth of STAT3-driven solid tumors through combined inhibition of JAK, SRC, and class III/V receptor tyrosine
kinases. Mol Cancer Ther. 2016;15:2334–43. Article CAS PubMed Google Scholar * Zheng L, Jia X, Zhang C, Wang D, Cao Z, Wang J, et al. Angiotensin II in atrial structural remodeling: the
role of Ang II/JAK/STAT3 signaling pathway. Am J Transl Res. 2015;7:1021–31. PubMed PubMed Central Google Scholar * Smith GS, Kumar A, Saba JD. Sphingosine phosphate lyase regulates
murine embryonic stem cell proliferation and pluripotency through an S1P(2)/STAT3 signaling pathway. Biomolecules. 2013;3:351–68. Article PubMed PubMed Central Google Scholar * Ying H,
Da L, Yu-xiu S, Yu X, Li-xia L, Li-mei X, et al. TLR4 mediates MAPK-STAT3 axis activation in bladder epithelial cells. Inflammation. 2013;36:1064–74. Article PubMed Google Scholar * Liu
BS, Cao Y, Huizinga TW, Hafler DA, Toes RE. TLR-mediated STAT3 and ERK activation controls IL-10 secretion by human B cells. Eur J Immunol. 2014;44:2121–9. Article CAS PubMed Google
Scholar * Lee YJ, Song H, Yoon YJ, Park SJ, Kim SY, Cho Han D, et al. Ethacrynic acid inhibits STAT3 activity through the modulation of SHP2 and PTP1B tyrosine phosphatases in DU145
prostate carcinoma cells. Biochem Pharm. 2020;175:113920. Article CAS PubMed Google Scholar * Baek SH, Lee JH, Ko JH, Lee H, Nam D, Lee SG, et al. Ginkgetin blocks constitutive STAT3
activation and induces apoptosis through induction of SHP-1 and PTEN tyrosine phosphatases. Phytother Res. 2016;30:567–76. Article CAS PubMed Google Scholar * Wilson HM. SOCS proteins in
macrophage polarization and function. Front Immunol. 2014;5:357. Article PubMed PubMed Central Google Scholar * Fang S, Liu B, Sun Q, Zhao J, Qi H, Li Q. Platelet factor 4 inhibits
IL-17/Stat3 pathway via upregulation of SOCS3 expression in melanoma. Inflammation. 2014;37:1744–50. Article CAS PubMed Google Scholar * Huttlin EL, Bruckner RJ, Navarrete-Perea J,
Cannon JR, Baltier K, Gebreab F, et al. Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell. 2021;184:3022–3040 e3028. Article CAS PubMed PubMed
Central Google Scholar * Zhu M, Wang Q, Gu T, Han Y, Zeng X, Li J, et al. Hydrogel-based microenvironment engineering of haematopoietic stem cells. Cell Mol Life Sci. 2023;80:49. Article
CAS PubMed PubMed Central Google Scholar * Miranda MB, McGuire TF, Johnson DE. Importance of MEK-1/-2 signaling in monocytic and granulocytic differentiation of myeloid cell lines.
Leukemia. 2002;16:683–92. Article CAS PubMed Google Scholar * Lu HC, Kim S, Steelman AJ, Tracy K, Zhou B, Michaud D, et al. STAT3 signaling in myeloid cells promotes pathogenic
myelin-specific T cell differentiation and autoimmune demyelination. Proc Natl Acad Sci USA. 2020;117:5430–41. Article CAS PubMed PubMed Central Google Scholar * Kumar V, Cheng P,
Condamine T, Mony S, Languino LR, McCaffrey JC, et al. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage
differentiation. Immunity. 2016;44:303–15. Article CAS PubMed PubMed Central Google Scholar * Korkmaz B, Moreau T, Gauthier F. Neutrophil elastase, proteinase 3 and cathepsin G:
physicochemical properties, activity and physiopathological functions. Biochimie. 2008;90:227–42. Article CAS PubMed Google Scholar * Savage COS. Vascular biology and vasculitis. Apmis.
2009;117:37–40. Article Google Scholar * Jones BE, Herrera CA, Agosto-Burgos C, Starmer J, Bass WA, Poulton CJ, et al. ANCA autoantigen gene expression highlights neutrophil heterogeneity
where expression in normal-density neutrophils correlates with ANCA-induced activation. Kidney Int. 2020;98:744–57. Article CAS PubMed PubMed Central Google Scholar * Sumi D, Suzukawa
K, Himeno S. Arsenic trioxide augments all-trans retinoic acid-induced differentiation of HL-60 cells. Life Sci. 2016;149:42–50. Article CAS PubMed Google Scholar * Klobuch S, Steinberg
T, Bruni E, Mirbeth C, Heilmeier B, Ghibelli L, et al. Biomodulatory treatment with azacitidine, all-trans retinoic acid and pioglitazone induces differentiation of primary AML blasts into
neutrophil like cells capable of ROS production and phagocytosis. Front Pharm. 2018;9:1380. Article CAS Google Scholar * Zhu H, Liu S, He W, Sun F, Li Y, Yang P, et al. One-step
genotyping method in loxP-based conditional knockout mice generated by CRISPR-Cas9 technology. Mol Biotechnol. 2022;64:1227–33. Article CAS PubMed PubMed Central Google Scholar * Kondo
Y, Ledderose C, Slubowski CJ, Fakhari M, Sumi Y, Sueyoshi K, et al. Frontline Science: Escherichia coli use LPS as decoy to impair neutrophil chemotaxis and defeat antimicrobial host
defense. J Leukoc Biol. 2019;106:1211–9. Article CAS PubMed Google Scholar * Chu Y, Chen Y, Guo H, Li M, Wang B, Shi D, et al. SUV39H1 regulates the progression of MLL-AF9-induced acute
myeloid leukemia. Oncogene. 2020;39:7239–52. Article PubMed PubMed Central Google Scholar * Huang D, Sun G, Hao X, He X, Zheng Z, Chen C, et al. ANGPTL2-containing small extracellular
vesicles from vascular endothelial cells accelerate leukemia progression. J Clin Investig. 2021;131:e138986. Article CAS PubMed PubMed Central Google Scholar * Zhu W, Liu Y, Zhang W,
Fan W, Wang S, Gu JH, et al. Selenomethionine protects hematopoietic stem/progenitor cells against cobalt nanoparticles by stimulating antioxidant actions and DNA repair functions. Aging.
2021;13:11705–26. Article CAS PubMed PubMed Central Google Scholar * Joshi HR, Hill HR, Zhou Z, He X, Voelkerding KV, Kumanovics A. Frontline science: Cxxc5 expression alters cell cycle
and myeloid differentiation of mouse hematopoietic stem and progenitor cells. J Leukoc Biol. 2020;108:469–84. Article CAS PubMed Google Scholar * Ko M, Huang Y, Jankowska AM, Pape UJ,
Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–43. Article CAS PubMed PubMed Central Google
Scholar * He J, Song Y, Li G, Xiao P, Liu Y, Xue Y, et al. Fbxw7 increases CCL2/7 in CX3CR1hi macrophages to promote intestinal inflammation. J Clin Investig. 2019;129:3877–93. Article
PubMed PubMed Central Google Scholar * Zhang J, Chen F, Li W, Xiong Q, Yang M, Zheng P, et al. 14-3-3zeta interacts with stat3 and regulates its constitutive activation in multiple
myeloma cells. PLoS One. 2012;7:e29554. Article CAS PubMed PubMed Central Google Scholar * Wu X, Liu H, Brooks A, Xu S, Luo J, Steiner R, et al. SIRT6 mitigates heart failure with
preserved ejection fraction in diabetes. Circ Res. 2022;131:926–43. Article CAS PubMed PubMed Central Google Scholar * Zhang S, Liu H, Yin M, Pei X, Hausser A, Ishikawa E, et al.
Deletion of protein kinase D3 promotes liver fibrosis in mice. Hepatology. 2020;72:1717–34. Article CAS PubMed Google Scholar * Xiao Z, Xu H, Qu ZY, Ma XY, Huang BX, Sun MS, et al.
Active ingredients of reduning injection maintain high potency against SARS-CoV-2 variants. Chin J Integr Med. 2023;29:205–12. Article CAS PubMed Google Scholar * N’Guessan K, Grzywa R,
Seren S, Gabant G, Juliano MA, Moniatte M, et al. Human proteinase 3 resistance to inhibition extends to alpha-2 macroglobulin. FEBS J. 2020;287:4068–81. Article PubMed Google Scholar *
Fujinaga M, Chernaia MM, Halenbeck R, Koths K, James MN. The crystal structure of PR3, a neutrophil serine proteinase antigen of Wegener’s granulomatosis antibodies. J Mol Biol.
1996;261:267–78. Article CAS PubMed Google Scholar * Chiou J, Lin YF, Hsiao M. Abstract A24: Translation control of tumor metastasis via FSTL1 binding with SPP1/OPN to modulated
cytoskeleton remodeling. Cancer Res. 2017;77(6_Supplement):A24. Download references ACKNOWLEDGEMENTS We thank Dr. Hongbo R. Luo and Xuemei Xie at the Department of Pathology, Harvard Medical
School, Children’s Hospital Boston, and Dana-Farber/Harvard Cancer Center, Boston, Massachusetts, USA. for suggestions and guidelines. We thank Dr. Tao Cheng, Dr. Hui Cheng, Dr. Weiping
Yuan and Dr. Guoguang Zheng at the State Key Laboratory of Experimental Hematology, National Clinical Research Center for Blood Diseases for the gift of significant experiment materials.
FUNDING This work was supported by the CAMS Innovation Fund for Medical Sciences (Grant No. 2021-12M-1-017), the National Natural Science Foundation of China (Grant No. 81970107, 82300249
and 82070192), and the Technology Development Project of Shandong Medicine and Health Science (202002050143). The funders had no role in the study design, data collection and analysis,
decision to publish or preparation of the manuscript. AUTHOR INFORMATION Author notes * These authors contributed equally: Huan Liu, Lu Sun. AUTHORS AND AFFILIATIONS * State Key Laboratory
of Experimental Hematology, National Clinical Research Center for Blood Diseases, Haihe Laboratory of Cell Ecosystem, Institute of Hematology & Blood Diseases Hospital, Chinese Academy
of Medical Sciences & Peking Union Medical College, Tianjin, 300020, China Huan Liu, Lu Sun, Hongfei Zhao, Zihan Zhao, Shiyue Zhang, Shan Jiang, Tianran Cheng, Tong Wang, Ya Shao, Yigeng
Cao, Erlie Jiang & Yuanfu Xu * Tianjin Institutes of Health Science, Tianjin, 301600, China Huan Liu, Lu Sun, Hongfei Zhao, Zihan Zhao, Shiyue Zhang, Shan Jiang, Tianran Cheng, Tong
Wang, Ya Shao, Yigeng Cao, Erlie Jiang & Yuanfu Xu * Clinical Laboratory, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, 710061, China Shan Jiang * The Second School
of Clinical Medicine, Guangdong Medical University, Dongguan, 523808, China Xiaohan Wang * Department of Clinical Lab, Weihai Municipal Hospital, Weihai, 264200, China Haiyan Zhu *
Department of Medical Laboratory, School of Clinical Medicine, Ningxia Medical University; Ningxia Key Laboratory of Clinical and Pathogenic Microbiology, General Hospital of Ningxia Medical
University, Yinchuan, 750001, China Huijuan Han * Hematopoietic Stem Cell Transplantation Center, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences
and Peking Union Medical College, Tianjin, 300020, China Yigeng Cao & Erlie Jiang * Department of Microbiology, Tumor and Cell Biology, Karolinska Institute, Solna, 17165, Sweden Yihai
Cao Authors * Huan Liu View author publications You can also search for this author inPubMed Google Scholar * Lu Sun View author publications You can also search for this author inPubMed
Google Scholar * Hongfei Zhao View author publications You can also search for this author inPubMed Google Scholar * Zihan Zhao View author publications You can also search for this author
inPubMed Google Scholar * Shiyue Zhang View author publications You can also search for this author inPubMed Google Scholar * Shan Jiang View author publications You can also search for this
author inPubMed Google Scholar * Tianran Cheng View author publications You can also search for this author inPubMed Google Scholar * Xiaohan Wang View author publications You can also
search for this author inPubMed Google Scholar * Tong Wang View author publications You can also search for this author inPubMed Google Scholar * Ya Shao View author publications You can
also search for this author inPubMed Google Scholar * Haiyan Zhu View author publications You can also search for this author inPubMed Google Scholar * Huijuan Han View author publications
You can also search for this author inPubMed Google Scholar * Yigeng Cao View author publications You can also search for this author inPubMed Google Scholar * Erlie Jiang View author
publications You can also search for this author inPubMed Google Scholar * Yihai Cao View author publications You can also search for this author inPubMed Google Scholar * Yuanfu Xu View
author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS HL and YX conceived the study; HL, LS, HZ, ZZ, SZ, SJ, TC, XW, TW, YS, HZ, HH, YC and EJ curated
the data; HL and LS formally analyzed the data; HL and LS performed the investigation; HL and LS determined the methodology; YX supervised the study; YX administered the project; HL, YC and
YX wrote, edited, and reviewed the manuscript. All authors read and approved the final manuscript. CORRESPONDING AUTHORS Correspondence to Yihai Cao or Yuanfu Xu. ETHICS DECLARATIONS
COMPETING INTERESTS The authors declare no competing interests. ETHICS APPROVAL Animal experiments were approved by our institutes and complied with the Guideline for Animal Care at the
State Key Laboratory of Experimental Hematology (IHCAMS-DWLL-CIFMS2021004-1). Human specimens were supplied by the Department of Hematology at the Institute of Hematology & Blood
Diseases Hospital. Written informed consent was obtained from the patients and approved by the Ethics Committee for Medical Research at the Department of Hematology at the Institute of
Hematology & Blood Diseases Hospital (CIFMS2021004-EC-E2). ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps
and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURE ORIGINAL DATA FILE RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original
author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the
article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use
is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Liu, H., Sun, L., Zhao, H. _et al._ Proteinase 3 depletion attenuates leukemia by
promoting myeloid differentiation. _Cell Death Differ_ 31, 697–710 (2024). https://doi.org/10.1038/s41418-024-01288-4 Download citation * Received: 04 December 2023 * Revised: 19 March 2024
* Accepted: 22 March 2024 * Published: 08 April 2024 * Issue Date: June 2024 * DOI: https://doi.org/10.1038/s41418-024-01288-4 SHARE THIS ARTICLE Anyone you share the following link with
will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative