Phc2 controls hematopoietic stem and progenitor cell mobilization from bone marrow by repressing vcam1 expression
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT The timely mobilization of hematopoietic stem and progenitor cells (HSPCs) is essential for maintaining hematopoietic and tissue leukocyte homeostasis. Understanding how HSPCs
migrate between bone marrow (BM) and peripheral tissues is of great significance in the clinical setting, where therapeutic strategies for modulating their migration capacity determine the
clinical outcome. Here, we identify an epigenetic regulator, Phc2, as a critical modulator of HSPC trafficking. The genetic ablation of _Phc2_ in mice causes a severe defect in HSPC
mobilization through the derepression of _Vcam1_ in bone marrow stromal cells (BMSCs), ultimately leading to a systemic immunodeficiency. Moreover, the pharmacological inhibition of VCAM-1
in _Phc2_-deficient mice reverses the symptoms. We further determine that Phc2-dependent _Vcam1_ repression in BMSCs is mediated by the epigenetic regulation of H3K27me3 and H2AK119ub.
Together, our data demonstrate a cell-extrinsic role for Phc2 in controlling the mobilization of HSPCs by finely tuning their bone marrow niche. SIMILAR CONTENT BEING VIEWED BY OTHERS CD63
ACTS AS A FUNCTIONAL MARKER IN MAINTAINING HEMATOPOIETIC STEM CELL QUIESCENCE THROUGH SUPPORTING TGFΒ SIGNALING IN MICE Article 06 August 2021 EPIGENETIC PROGRAMMING DEFINES HAEMATOPOIETIC
STEM CELL FATE RESTRICTION Article 01 May 2023 CLONAL HEMATOPOIESIS RELATED TET2 LOSS-OF-FUNCTION IMPEDES IL1Β-MEDIATED EPIGENETIC REPROGRAMMING IN HEMATOPOIETIC STEM AND PROGENITOR CELLS
Article Open access 07 December 2023 INTRODUCTION Hematopoietic stem and progenitor cells (HSPCs) are rare populations of cells that can sustain the hematopoietic system by continuously
replenishing blood cells1,2. Postnatally, most HSPCs engraft and reside in the BM niche, but a small fraction of these cells is mobilized and migrate into the peripheral blood (PB) to
reconstitute the hematopoietic system under specific signaling cues1,3,4. Therefore, identifying key regulators that critically control HSPC mobilization and understanding their underlying
mechanisms have been of significant interest to facilitate BM transplantation efficiency and to therapeutically inhibit the malignant migration of the transformed hematopoietic cells into
peripheral tissues5. Indeed, several BM niche proteins, including stem cell factor (kit ligand), CXCL12 (SDF-1α), and β1 integrin have been identified as key signaling components to control
the maturation of HSPCs, their migration into the circulation and their homing to the BM by mediating the interaction between HSPCs and bone marrow stromal cells (BMSCs)2,5,6,7. However, the
molecular pathways that regulate the expression of these BM niche proteins remain unclear. Polycomb group (PcG) proteins function as transcriptional repressors of target genes by mainly
modulating histone methylation8. These PcG proteins can be divided into two functionally distinct complexes: polycomb repressor complex 1 (PRC1) and polycomb repressor complex 2 (PRC2). PRC2
induces the formation of trimethylated histone H3 at lysine 27 (H3K27me3) through methyltransferase activity of Ezh proteins which are component proteins in PRC2 complex9. The canonical
PRC1 then recognizes and binds to H3K27me3 to sustain the transcriptional repression of a target gene8,10,11. Both canonical and noncanonical PRC1 complexes can cause additional
transcriptional silencing as E3 ubiquitin ligases by forming monoubiquitylated histone H2A at lysine 119 (H2AK119ub)8,12,13. In this study, we demonstrate that Phc2, a component of the
canonical PRC1, regulates HSPC mobilization through the repression of _Vcam1_ expression by enhancing both H3K27me3 and H2AK119ub in BMSCs. Therefore, Phc2 deficiency causes a severe HSPC
mobilization defect via the derepression of _Vcam1_ in BMSCs, and the pharmacological inhibition of VCAM-1 in BMSCs significantly reverses the symptoms of Phc2-deficient mice. These data
demonstrate the critical cell-extrinsic role of Phc2 in controlling HSPC mobilization and provide the first evidence of epigenetic control over HSPC mobilization. RESULTS PHC2 DEFICIENCY
LEADS TO A DEFECT IN HSPC MOBILIZATION As an initial step to elucidate the functional role of Phc2 during hematopoiesis, we characterized immune phenotypes of _Phc2_−/− (KO) mice14 compared
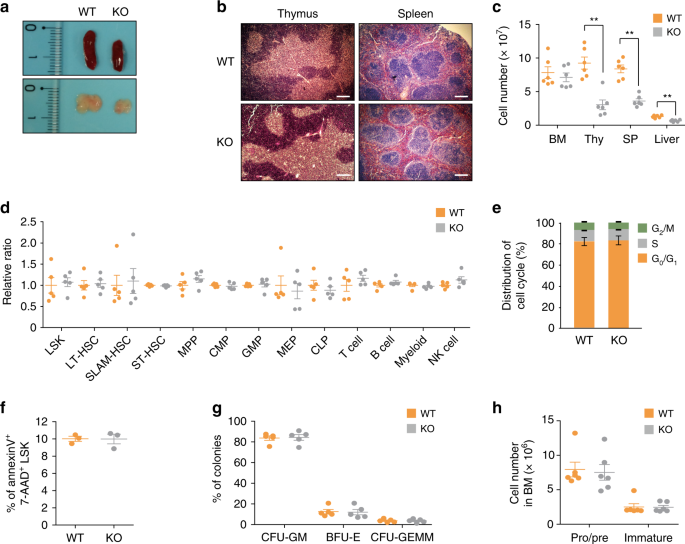
to those of _Phc2_+/+ (wild-type, WT) mice. We observed macroscopic abnormalities in the thymus and peripheral lymphoid organs of KO mice compared to those of WT mice. Both the thymus and
spleen of the KO mice maintained normal architecture, but these organs were smaller than those of the WT mice (Fig. 1a, b). Consistent with this observation, the absolute numbers of immune
cells and their precursors in the PB, thymus, spleen, and liver of the KO mice were significantly reduced compared to those of the WT mice (Tables 1 and 2, Fig. 1c). Notably, the absolute
number of early T-cell precursors (ETPs) in the thymus or immature B cells in the spleen was significantly lower in the KO mice relative to the WT mice (Table 1). Unexpectedly, the absolute
numbers and relative ratios of HSPCs, early progenitor cells, and immune cells in the BM were similar among all examined mice (Table 1 and Fig. 1c, d). Additionally, the loss of _Phc2_ did
not influence the cell cycle progression or apoptosis of HSPCs (Fig. 1e, f), and both the WT and KO BM-resident HSPCs exhibited no difference in terms of their ability to generate
multipotential or myeloid progenitor cells (Fig. 1g). The absolute numbers of WT and KO B cells in the BM at each developmental stage were not significantly different either (Fig. 1h). Given
that no functional defect was evident in the HSPCs of the BM but that there was a shortage of ETPs and immature B cells from the BM, we postulated that a Phc2 deficiency could lead to a
systemic immunodeficiency due to a defect in HSPC migration into the circulation. To test this hypothesis, we examined the numbers of circulating HSPCs from WT and KO mice using a
colony-forming unit (CFU) assay. The absolute numbers of clonogenic progenitors in the PB and spleen were decreased by approximately half in the KO mice compared to the WT mice, whereas the
absolute numbers of clonogenic progenitors in the BM were comparable between the WT and KO mice (Supplementary Fig. 1). To confirm this result, we performed in vivo HSPC mobilization assays
using two commonly used mobilization regimens for therapy, granulocyte colony-stimulating factor (G-CSF) and AMD3100 (CXCR4 antagonist)15,16,17,18. Five days after G-CSF treatment, the
absolute numbers of white blood cells (WBCs) and splenocytes from the KO mice remained significantly lower than those from the WT mice (Fig. 2a, b). The spleen size of the KO mice also
remained much smaller than that of the WT mice after G-CSF treatment (Fig. 2b). Consistent with these results, both the frequencies and absolute numbers of Lin−Sca-1+c-kit+ cells (LSK cells)
in the PB and spleen were significantly lower in the KO mice than in the WT mice (Supplementary Fig. 2). Likewise, the absolute numbers of clonogenic progenitors in the PB and spleen of the
KO mice were significantly reduced compared to those of the WT mice (Fig. 2c). However, the frequencies and absolute numbers of LSK cells in the BM were comparable between the WT and KO
mice (Supplementary Fig. 2). Moreover, the absolute numbers of clonogenic progenitors in the BM were not significantly different between the WT and KO mice (Fig. 2c). When we used an AMD3100
as a mobilizing agent of LSK, we still observed that the migration of KO LSK into the periphery is statistically decreased compared with that of WT LSK (Fig. 2d, e). However, the defective
migration of KO LSK after treatment of AMD3100 is relatively modest when we compared with results from G-CSF treatment (Fig. 2d, e). To remove circulating HSPCs and visualize neo-HSPC
mobilization from BM, we further treated WT and KO mice with 5-fluorouracil (5-FU), a chemotherapeutic agent that depletes circulating HSPCs and promotes HSPC release from BM during
hematopoietic repopulation19,20. The absolute numbers of WBCs and splenocytes recovered from KO mice were lower than those recovered from WT mice starting on day 8 after 5-FU treatment (Fig.
2f, g). The most prominent differences in the absolute numbers of WBCs and splenocytes or the size of the spleen between WT and KO mice was observed on day 16 after 5-FU treatment (Fig. 2f,
g). On day 16 after 5-FU treatment, the frequency and the absolute number of recovered LSK cells and the absolute number of recovered clonogenic progenitors in the PB and spleen were
significantly lower in KO mice than in WT mice, whereas those in the BM were comparable between the WT and KO mice (Fig. 2h, Supplementary Fig. 3). Taken together, these results indicate
that Phc2 deficiency causes a defect in HSPC mobilization from the BM into the periphery. _PHC2_ _−/_− BMSCS CAUSE A DEFECT IN HSPC MOBILIZATION To identify which cell type in the BM niche
contributed to the defect in HSPC release into the circulation of KO mice, we adoptively transferred LSK cells isolated from WT CD45.1 mice into irradiated WT or KO CD45.2 recipient mice
(Fig. 3a). KO recipients showed impaired repopulation of donor-derived immune cells in the thymus and spleen (Fig. 3b, c). However, the absolute numbers of LSK cells and lineage-committed
progenitors (Lin−c-kit+ cells; LK cells) in the BM of KO recipients were comparable to those in the WT recipients (Fig. 3b–d). To confirm these observations, irradiated WT CD45.1 mice were
reconstituted with LSK cells isolated from WT or KO CD45.2 mice (Fig. 3e). The origin of the donor LSK cells did not affect the repopulation of leukocytes in WT recipients (Fig. 3f, g). The
origin of the donor LSK cells did not affect the absolute number of LSK cells or LK cells in the BM of WT recipients either (Fig. 3h). We also performed a serial competitive BM repopulation
assay to further determine whether KO HSPCs had an intrinsic defect. We found that KO HSPCs had the same ability to reconstitute immune cells as WT HSPCs (Supplementary Fig. 4). This
strongly suggests that the defect in HSPC release into the circulation in KO mice is caused by an extrinsic factor from BMSCs within BM niches. To confirm this result, we performed an in
vivo homing assay with CFSE-labeled WT and KO LSK cells. The number of CFSE+ LSK cells homing to the BM was higher among the KO recipients than among the WT recipients (Fig. 4a). Such an
increase in the short-term migration of CFSE+ LSK cells into the BM of KO recipients led to fewer CFSE+ LSK cells homing to the PB and spleen of the KO recipients (Fig. 4a). Accordingly, the
frequency of clonogenic progenitors in the BM of the KO recipients was increased, whereas the frequencies of clonogenic progenitors in the PB and spleen of the KO recipients were decreased
compared to those of the WT recipients (Fig. 4b, Supplementary Fig. 5). We next measured the capacity of WT and KO LSK cells to transmigrate across their BMSCs using a trans-stromal
migration assay. The number of LSK cells transmigrating across KO BMSCs was smaller than the number of LSK cells transmigrating across WT BMSCs (Fig. 4c, Supplementary Fig. 6). In contrast,
the number of LSK cells adhering to KO BMSCs was greater than the number of LSK cells adhering to WT BMSCs (Fig. 4d, Supplementary Fig. 6). Collectively, these data demonstrate that an
extrinsic factor from BMSCs causes HSPCs to adhere more strongly to BMSCs, ultimately leading to the defective mobilization of HSPCs from the BM into the periphery in KO mice. PCG PROTEINS
REPRESS _VCAM1_ GENE EXPRESSION IN BMSCS To understand the molecular mechanisms through which PcG strengthens the adhesion properties of HSPCs to BMSCs, we first investigated the
compositions and architectures of BM niches in WT and KO mice and measured the expression levels of candidate genes that are known to be involved in these processes. Endothelial cells,
mesenchymal stem cells, and osteoblasts are major BMSCs found in BM niches21,22,23. Importantly, neither a change in BMSC composition nor an abnormal architecture was found in the KO BM
niches (Supplementary Figs. 7 and 8). Next, we examined the expression patterns of soluble factors and cell surface molecules that might regulate HSPC mobilization in WT and KO BMSCs2,7
(Fig. 5a, Supplementary Fig. 9). Intriguingly, both the mRNA and protein levels of VCAM-1 were significantly increased in KO BMSCs compared to WT BMSCs (Fig. 5a, b). Flow cytometry analysis
further confirmed that the expression of VCAM-1 was significantly higher in KO BMSCs than in WT BMSCs (Fig. 5c). Consistent with this observation, _Vcam1_ mRNA levels were significantly
elevated in activated KO peritoneal macrophages compared to those of WT (Supplementary Fig. 10). Taken together, these results indicate that Phc2 may repress _Vcam1_ expression in BMSCs. To
further examine Phc2-dependent _Vcam1_ repression, we knocked down Phc2 expression in BMSCs (OP9 cells) and concurrently monitored _Vcam1_ expression levels under treatment with TNF-α, a
well-known inducer of _Vcam1_ transcription24 (Fig. 5d). As expected, TNF-α treatment induced the expression of _Vcam1_ at 3 h post-treatment. However, this was significantly exacerbated
when Phc2 was depleted, and thus, the VCAM-1 level was not dampened up to 12 h post-TNF-α treatment (Fig. 5e, f). Overall, these results suggest that Phc2 represses the expression of _Vcam1_
in BMSCs at the transcriptional level. To demonstrate a role for Phc2 in the transcriptional repression of _Vcam1_, we performed a chromatin immunoprecipitation (ChIP) assay with seven
primer pairs covering the putative Phc2 binding sites located in the _Vcam1_ gene (Fig. 6a). This analysis revealed the strong binding of Phc2 to the promoter region of the _Vcam1_ locus
(Fig. 6b, primers #2 and #3). Interestingly, direct binding by other members of the canonical PRC1 family, such as Bmi1, Ring1b, and Cbx7, was also found around the same region covered by
primers #2 and #3 (Fig. 6c). Consistent with this, Bmi1, Ring1b, and Cbx7 binding to these regions were significantly decreased in the absence of Phc2 (Fig. 6c). The acetylation of histone
H3 at lysine 9/14 (H3K9ac), trimethylation of histone H3 at lysine 4 (H3K4me3), H3K27me3, and H2AK119ub were observed around the same region covered by primers #2 and #3 (Fig. 6c). H3K9ac
and H3K4me3 were increased, whereas H3K27me3 and H2AK119ub were decreased in KO BM cells compared to WT BM cells (Fig. 6c). Furthermore, the treatment of BMSCs (OP9 cells) with GSK126, an
Ezh2 inhibitor25, led to the upregulation of _Vcam1_ expression in a dose-dependent manner (Fig. 6d, e and Supplementary Fig. 11). Consistent with these observations, the binding of Phc2,
Bmi1, and Ring1b to the regions covered by primers #2 and #3 was decreased in GSK126-treated BMSCs (OP9 cells) (Fig. 6f). Next, we then asked whether GSK126 treatment could mimic Phc2
deficiency in vivo. Different dose of GSK126 was administered intraperitoneally into WT mice. Sixteen hours after GSK injection, BMSCs were isolated and analyzed for VCAM-1 expression. We
also measured the capacity of WT LSK cells to transmigrate across their GSK126-treated BMSCs using a trans-stromal migration assay. Results clearly showed that GSK126 treatment increased
both VCAM-1 expression in BMSCs and adherence of LSKs onto BMSCs in a dose-dependent manner (Supplementary Fig. 12). Therefore, the canonical PRC1 containing Phc2 can repress _Vcam1_
expression through histone modifications, such as by enhancing H3K27me3 and H2AK119ub, in BMSCs. ANTI-VCAM-1 AB TREATMENT RESTORES _PHC2_ _−/_− HSPC MOBILIZATION Next, we examined whether
blocking the interaction between VCAM-1 and VLA-4 could restore HSPC mobilization in KO mice. We first attempted to measure the mobilization capacity of WT LSK cells across KO BMSCs after
treatment with neutralizing antibodies (Abs) against VCAM-1 and/or VLA-4. Treatment with either Ab restored the trans-stromal migration activity of LSK cells across KO BMSCs to a level
similar to that of LSK cells across WT BMSCs (Fig. 7a). Additionally, there was no significant difference in the relative ratio of LSK cell adhesion to KO or WT BMSCs after treatment with
the neutralizing Abs (Fig. 7b). The in vivo homing assay further confirmed that blocking the interaction between VCAM-1 and VLA-4 could restore LSK cell mobilization from the KO BM into the
PB and spleen (Fig. 7c, d and Supplementary Fig. 13). We then tested whether treatment of the neutralizing VCAM-1 Ab could rescue steady-state HSPC mobilization in KO mice. Results clearly
demonstrated that the administration of the neutralizing VCAM-1 Ab in KO mice successfully reconstitutes HSPC mobilization from BM into periphery (Fig. 7e–g). We also performed a G-CSF-,
AMD3100-, or 5-FU-induced LSK cell mobilization assay using WT and KO mice treated with the neutralizing VCAM-1 Ab. There were no significant differences in the absolute numbers of WBCs and
splenocytes between the WT and KO mice treated with anti-VCAM-1 on day 5 after the G-CSF treatment and on day 16 following the 5-FU challenge (Fig. 8a, b, f, g). Similarly, the absolute
number of LSK cells and the clonogenic progenitor frequency in the BM, PB, and spleen were comparable between WT and KO recipients treated with the neutralizing Ab on day 5 after the G-CSF
treatment and on day 16 following the 5-FU challenge (Fig. 8c, h, Supplementary Figs. 14 and 15). Consistent with these results, the defective HSPC migration into the PB of the KO mice was
reversed by treating the mice with anti-VCAM-1, as observed in the HSPC mobilization assay using AMD3100 alone or in combination with G-CSF (Fig. 8d, e and Supplementary Fig. 16). To further
reinforce these observations, we assessed the significance of VCAM-1 blockage when the migration defect of HSPCs are driven by extrinsic microenvironment which mimics the phenotype
associated with Phc2 KO mice. First, we adoptively transferred LSK cells isolated from WT CD45.1 mice into irradiated WT or KO CD45.2 recipient mice and then performed the same G-CSF-induced
LSK cell mobilization assay using recipient mice treated with the neutralizing VCAM-1 Ab (Supplementary Fig. 17). Consistent to the HSPC mobilization assay in KO mice, WT cells
reconstituted in irradiated KO recipient cells were only partially rescued the mobilization defects when the single G-CSF agent was treated (Supplementary Fig. 17). However, the
administration of the neutralizing VCAM-1 Ab in combination with G-CSF in KO recipient mice completely reconstituted WT donor HSPC mobilization from BM into periphery, the same degree to WT
recipient mice (Supplementary Fig. 17). Taken together, these results demonstrate that blocking the interaction between VCAM-1 and VLA-4 can restore HSPC migration from the BM into the
periphery of KO mice. DISCUSSION In this study, we demonstrated that Phc2 binds to the _Vcam1_ locus to repress its transcription by enhancing H3K27me3 and H2AK119ub in BMSCs. In the absence
of Phc2, the increased _Vcam1_ expression in BMSCs strengthened the interaction between HSPCs and BMSCs, compromising timely HSPC mobilization from the BM into the periphery, which in turn
led to a systemic immunodeficiency. Consistent with these observations, treatment with a neutralizing Ab against VCAM-1 in Phc2-deficient mice restored HSPC mobilization from the BM into the
periphery. These data firmly establish a causative role for the epigenetic regulation of VCAM-1 in regulating HSPC mobilization, as a key downstream mediator of Phc2 functions. Recent
genetic evidence from conditional VCAM-1- or VLA-4-deficient mice further supports our model of the VLA-4/VCAM-1 pathway as downstream of Phc2 function, and as expected from our results with
Phc2-deficient mice overexpressing VCAM-1, VCAM-1- or VLA-4 deficient mice exhibit increased circulating progenitors and immature B cells, and impaired HSPC homing to BM26,27,28,29,30. The
BM homing defect of HSPCs in these mice became obvious when the mice were treated with 5-FU or G-CSF26,27. However, the Phc2-deficient mice still displayed the migration defect of HSPCs from
the BM into the periphery after treatment with these mobilization agents. The identification of a single key mediator of Phc2 function may have important clinical implications. Similar to
previous observations with conditional VCAM-1- or VLA-4-deficient mice26,27,28,29,30, we found an abnormality of only steady-state HSPC mobilization in Phc2-deficient mice. Additionally,
treatment with the anti-VCAM-1 Ab or anti-VLA-4 Ab in primates or mice does not cause any other physiological change31,32. Combined evidence may suggest a dominant role for VCAM-1 in
steady-state HSPC migration due to the VCAM-1 expression pattern. In steady-state conditions, the BMSC is the only cell type that constitutively expresses VCAM-1 at physiological levels,
although vascular endothelial cells and other immune cell populations express minimally detectable levels of VCAM-133,34,35,36,37,38. Furthermore, with a proof-of-concept experiment, we
successfully demonstrated that blocking VCAM-1 in BM niches critically reverses the HSPC mobilization defect in Phc2-deficient mice. Therefore, along with other recent data39,40,41,42,43,
our results suggest that modulating the interaction between VCAM-1 and VLA-4 in BM niches provides potential strategies to develop HSPC mobilizing agents or anti-leukemia drugs. Therefore,
it will be interesting to observe whether a specific inhibitor that blocks Phc2 function is useful for keeping malignant cells from hematopoietic lineages within the BM. To our knowledge, we
are the first to provide evidence that a systemic immunodeficiency can be caused by a defect in a particular epigenetic regulator that represses the expression of a certain cell adhesion
molecule in BMSCs. Our study also demonstrates the molecular mechanism through which the epigenetic regulation of extrinsic factors can help finely tune the interaction between HSPCs and
BMSCs within BM niches during HSPC mobilization. Although several genes that are involved in hematopoiesis have been identified as targets for epigenetic regulation, the epigenetic effect on
HSPC mobilization has not been elucidated. Notably, the vast majority of previously identified genes play important roles in defining the intrinsic property of HSCs and the development of
leukemia8,44,45. Like deficiencies in other epigenetic regulators, a single PcG gene deficiency in mice mostly causes early embryonic lethality or a limited life span with severe
developmental defects, including an intrinsic defect in HSCs8,46,47. This characteristic hinders the search for additional roles of polycomb proteins in the regulation of immune phenomena
aside from their known functions in HSC differentiation. Similar to other members of the PcG protein family, Phc2 is expressed in various tissues and cells and regulates the transcription of
_Cdkn2a_ (_p16__INK4a_ and _p19__ARF_) and _hox_ genes through direct associations with chromatin8,14. However, Phc2-deficient mice are fertile and exhibit less severe defects associated
with the abnormal axial skeleton and Peyer’s patch14,48. Moreover, the systemic immunodeficiency observed in Phc2-deficient mice is not caused by an intrinsic defect in HSC function, but
instead, the Phc2 deficiency causes a defect in HSPC mobilization by enhancing the expression of VCAM-1 in BMSCs. This may suggest that most of the Phc2 functions in mammals are dispensable
or can be replaced by other components of the canonical PRC1 due to a structural heterogeneity of PRC1 or functional redundancy of PcG proteins. In fact, each of the mammalian PRC2 or PRC1
subunits has several paralogs8,9,10,11,12,13. For example, Phc2 has two paralogs, Phc1 and Phc38,9,10,11,12,13. Previous observation revealed that Phc1 deficiency causes a defect in HSC’s
self-renewal activity49,50. Therefore, Phc1, but not phc2, might be critical for HSC maintenance and function. Consistent with this idea, the expression and epigenetic pattern of _Cdkn2a_
(_p16__INK4a_ and _p19__ARF_) in KO BM cells showed no significant differences compared to those in WT BM cells (Supplementary Fig. 18). Another example of nonredundant function by each
paralog within PRC2 or PRC1 components might be found in Ezh proteins or Cbx proteins, respectively51,52,53,54,55. Among Ezh proteins, Ezh1 is critical for self-renewal activity of HSCs,
whereas Ezh2 is dispensable for proper HSC function51,52. Also, Cbx7 is the only paralog which is necessary for self-renewal activity of HSC among Cbx proteins53,54. In our molecular model,
PRC2 containing Ezh2 with methyltransferase activity initiates H3K27me3 on the _Vcam1_ locus in BMSCs because the Ezh2 inhibitor (GSK126) treatment of BMSCs increases the _Vcam1_ mRNA levels
by negating the recognition of H3K27me3 by the canonical PRC1 complex containing Phc2 on the same locus. The canonical PRC1 then recognizes and binds to H3K27me3 to maintain _Vcam1_ gene
suppression. The recognition of H3K27me3 on the _Vcam1_ locus by the canonical PRC1 is dependent on _Phc2_ expression. Consistently, we observed reductions in the binding of other canonical
PRC1 components to the _Vcam1_ locus when Phc2 was not expressed. Our study also revealed that the H2AK119ub activity of the canonical PRC1 on the _Vcam1_ locus was dependent on _Phc2_
expression, which is consistent with a recent observation that the interaction between Bmi1 and Phc2 is critical for the H2AK119ub activity of the canonical PRC155. In conclusion, the
results of our study demonstrated Phc2-regulated HSPC mobilization through the direct repression of _Vcam1_ gene expression in BMSCs. These findings may offer a novel approach to
manipulating the epigenetic patterns of genes involved in HSPC mobilization, leading to new therapeutic strategies for BM transplantation and leukemia treatment. METHODS EXPERIMENTAL MICE
AND PHENOTYPIC ANALYSIS _Phc2_+/− (_Phc2_ heterozygote) mice with a C57BL/6 background were used in this study14. C57BL/6 and B6 CD45.1 mice were obtained from The Jackson Laboratory. WT and
KO mice were bred from _Phc2_+/− mice. All animals received proper care in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The study
protocol was approved by the Institutional Animal Care and Use Committee of Korea University (protocol numbers: KUIACUC-20110104-3, KUIACUC-20141002-4, and KUIACUC-20160927-3). CELL CULTURE
BMSCs were obtained and maintained from WT and KO mice56. Briefly, bone marrow (BM) cells were isolated from the femurs and tibias of 8-week-old mice and cultured in Dulbecco’s modified
Eagle medium (Welgene LM 001-05) supplemented with 10% fetal bovine serum (FBS; Welgene S 001–01), 10% horse serum (Welgene S 104–01), 2 mM L-glutamine (Welgene LS 002–01), 2 mM sodium
pyruvate (Welgene LS 002–02), and 100 U ml−1 penicillin/streptomycin (Welgene LS 202–02) at 37 °C and 5% CO2. Non-adherent cells were removed by washing with PBS after overnight incubation.
Adherent cells were cultured and fresh medium was added every 2 days. After 4 weeks of culture, cells were harvested by incubation with trypsin/EDTA (Welgene LS 015–01) for 2 min at 37 °C
and plated onto culture dishes. At two weeks after the first passage, cells were harvested and cultured under the same conditions for subsequent passages (passages 2–7). OP9 BMSC line (ATCC
CRL-2749) was maintained in alpha MEM (Welgene LM 008-02) supplemented with 20% FBS and 1% penicillin/streptomycin at 37 °C and 5% CO2. For induction of VCAM-1, OP9 cells were treated with
TNF-α (100 ng ml−1; R&D Systems 410-MT) for the indicated time period. To inhibit Ezh activity, OP9 cells were treated with various concentrations of GSK126, an Ezh2 inhibitor (Xcessbio
Biosciences M60071-2), for 48 h. PB COUNT, FLOW CYTOMETRY ANALYSIS, AND CELL SORTING PB samples from each mouse were collected by retro-orbital bleeding and analyzed using a Coulter LH 780
Hematology Analyzer (Beckman Coulter). For flow cytometry, single cell suspensions were prepared from the BM, PB, thymus, and spleen of each mouse. After removing red blood cells, cells were
stained with MACS buffer at 4 °C in the presence of Fc Block Ab (BD Biosciences 553141, dilution 1:100). Abs purchased from BD Biosciences, Biolegend or Thermo Fisher Scientific were used
to detect the following cell surface markers by flow cytometry: B220 (BD Biosciences 553090, dilution 1:100), CD3ε (Thermo Fisher Scientific 11-0031-63, dilution 1:100), CD4 (BD Biosciences
553729, dilution 1:100), CD8 (BD Biosciences 553030, dilution 1:100), CD11b (Thermo Fisher Scientific 11-0112-41, dilution 1:100), CD11c (BD Biosciences 553802, dilution 1:100), CD16:32
(Thermo Fisher Scientific 14-0161-81, dilution 1:100), CD25 (Thermo Fisher Scientific 17-0251-81, dilution 1:100), CD31 (BD Biosciences 561814, dilution 1:100), CD34 (BD Biosciences 560238,
dilution 1:100), CD44 (BD Biosciences 561861, dilution 1:100), CD45 (BD Biosciences 550539, dilution 1:100), CD48 (Biolegend 103412, dilution 1:100), CD51 (BD Biosciences 551187, dilution
1:100), CD106 (BD Biosciences 553332, dilution 1:100), CD127 (Thermo Fisher Scientific 45-1271-80, dilution 1:100), CD150 (Biolegend 115922, dilution 1:100), c-kit (Thermo Fisher Scientific
17-1171-81, dilution 1:100), F4/80 (Thermo Fisher Scientific MF48004, dilution 1:100), Flt3 (Thermo Fisher Scientific 46-1351-80, dilution 1:100), IgD (BD Biosciences 562022, dilution
1:100), IgM (BD Biosciences 550676, dilution 1:100), NK1.1 (BD Biosciences 557391, dilution 1:100), and Sca-1 (BD Biosciences 558162, dilution 1:100). After washing several times with PBS,
stained cells were resuspended in PBS and analyzed by flow cytometry using a FACSCalibur with CellQuest software (BD Biosciences). Analysis and sorting of HSPCs (Lin–Sca-1+c-kit+ cells; LSK
cells) and early progenitor cells were performed using MACS cell separation system and FACSAria Fusion Cell Sorter (BD Biosciences)57. Depletion of specific lineage cells was achieved with a
lineage-specific Ab cocktail and anti-biotin microbeads (Miltenyi Biotec 130-090-858, dilution 1:5). Gating strategy for LSK cell sorting58 was depicted in Supplementary Fig. 19.
HISTOLOGICAL ANALYSIS AND IMMUNOBLOTTING For histological analysis59, tissue samples were fixed with 10% formalin (Sigma, HT5011) and embedded in paraffin. Then, sections measuring 5 μm were
cut using a Leica CM1800 cryostat (Leica Microsystems), and air dried at room temperature. After dry, slides containing each tissue sample were stained with hematoxylin (Merck 1.05174.0500)
and eosin (Merck 109844) per the manufacturer’s protocol. For immunoblot analysis59, cells were lysed with RIPA lysis buffer containing 50 mM Tris, pH 7.4, 150 mM NaCl, 1% NP-40 (Sigma,
74385), 0.5% sodium deoxycholate (Sigma, D6750), 0.1% SDS (Sigma, L4509) with 200 μg ml−1 of phenylmethylsulfonyl fluoride (Sigma, P7626), phosphatase inhibitor cocktail (Sigma, P0044) and
protease inhibitor cocktail (Millipore, 535140). The cell lysates were then resolved by 12% SDS-polyacrylamide gel electrophoresis, transferred onto Immobilon P membranes (Millipore,
IPVH00010), and immunoblotted with anti-VCAM-1 Ab (Santa Cruz Biotechnology, Inc. sc-8304, dilution 1:1000), anti-H3K27me3 Ab (EMD Millipore 07-449, dilution 1:1000), or anti-histone H3 Ab
(Cell Signaling Technology 9715, dilution 1:1000) coupled with goat anti-rabbit IgG-HRP (Santa Cruz Biotechnology, Inc. sc-2004, dilution 1:2500), and anti-Phc2 Ab (mouse IgG, dilution
1:200)14 or anti-β-actin Ab (Sigma-Aldrich A5441, dilution 1:25,000) coupled with goat anti-mouse IgG-HRP (Santa Cruz Biotechnology, Inc. sc-2005, dilution 1:2500). Immunoreactive bands were
visualized using an ECL solution (Thermo Fisher Scientific 34080). To quantify the relative protein expression, immunoreactive bands were normalized to levels of β-actin. The relative band
intensity of protein expression was quantified using ImageJ software (National Institutes of Health). Uncropped blots can be found in the Source Data. CELL CYCLE ANALYSIS AND MEASUREMENT OF
APOPTOSIS BM-resident LSK cells from 8-week-old mice were isolated and incubated in 70% ethanol overnight at 4 °C. After washing with PBS, cells were stained with propidium iodide
(Sigma-Aldrich P4170) for cell cycle analysis. To measure apoptosis, LSK cells were stained with 7-AAD (BD Biosciences 559925, dilution 1:20) and Annexin V-FITC (BD Biosciences 556547,
dilution 1:20). After washing with PBS, stained cells were analyzed by flow cytometry. CFU ASSAY Mononuclear cells isolated from the BM, PB, and spleen were plated onto Methocult GF M3434
(StemCell Technologies 03444) in 35 mm cell culture dishes. Cells were then incubated at 37 °C and 5% CO2. After 7 days of incubation, type and number of colonies were determined60. IN VIVO
HSPC MOBILIZATION ASSAYS For G-CSF-induced mobilization assays15, recombinant human G-CSF (Amgen Filgrastim Neupogen) was diluted in PBS with 0.1% BSA (GenDEPOT A0100-050). G-CSF or vehicle
was administered by daily subcutaneous injection to 8-week-old mice at a dose of 250 μg kg−1 day−1 for 5 days. For AMD3100-induced mobilization assays17, AMD3100 (Sigma-Aldrich A5602) was
diluted in PBS and administered subcutaneously to 8-week-old mice at 5 mg kg−1. For G-CSF and AMD3100-induced mobilization assays61, G-CSF or vehicle was administered by daily subcutaneous
injection to 8-week-old mice at a dose of 250 μg kg−1 day−1 for 5 days. One h before final G-CSF dose, AMD3100 administered subcutaneously to the same group of mice at 5 mg kg−1. Three h
after the final G-CSF or AMD3100 dose, mice were sacrificed to assess the absolute numbers of nucleated cells in the PB and spleen. The absolute numbers of LSK cells in the BM, PB, and
spleen were determined by flow cytometry and CFU assays. To block the VLA-4 and VCAM-1 interaction, anti-VCAM-1 (BD Biosciences 553330) Ab or the respective isotype control (2 mg kg−1 day−1;
BD Biosciences 553926) was administered intravenously every day. For 5-FU-induced mobilization assays62, 5-FU (Sigma-Aldrich F6627) was administered intravenously to 8-week-old mice at 200
mg kg−1. The absolute numbers of nucleated cells in the PB and spleen were counted on days 4, 8, 12, and 16 after 5-FU treatment. On day 16 after 5-FU administration, mice were sacrificed,
and the absolute numbers of Lin−Sca-1+c-kit+ cells (LSK cells) in the BM, PB, and spleen were determined by flow cytometry and CFU assays. To block the VLA-4 and VCAM-1 interaction, mice
were injected with anti-VCAM-1 Ab or the respective isotype control (2 mg kg−1 day−1) from day 8 to day 15 after 5-FU administration. For anti-VCAM-1 Ab-induced mobilization assays63,
anti-VCAM-1 Ab or the respective isotype control administered by daily subcutaneous injection to 8-week-old mice at a dose of 2 mg kg−1 day−1 for 3 days. Eight hours after the final Ab dose,
mice were sacrificed, and the absolute numbers of nucleated cells in the PB and spleen were counted. The absolute numbers of LSK cells in the BM, PB, and spleen were determined by CFU
assays described above. LSK TRANSPLANTATION LSK cells (1 × 105) isolated from 8-week-old donor mice were injected intravenously into lethally irradiated (10 Gy) 8-week-old recipient mice.
Twelve weeks after the LSK transfer, recipient mice were sacrificed and analyzed for reconstitution of donor immune cells. Donor cells and recipient cells were discriminated by flow
cytometry using anti-CD45.2 (104)-PerCP-Cy5.5 Ab (BD 552950, dilution 1:100) for congenic strain (CD45.1) discrimination. For serial competitive LSK repopulation assays58, LSK cells (5 ×
104) isolated from WT or KO mice (CD45.2) were mixed with an equal number of LSK cells isolated from competitor mice (WT in CD45.1). The cell mixture was then intravenously injected into
lethally irradiated (10 Gy) 8-week-old recipient mice (WT in CD45.1). Secondary transplantation was performed at 12 weeks after primary engraftment. LSK cells (1 × 105) harvested from
primary transplants were intravenously injected into lethally irradiated (10 Gy) 8-week-old recipient mice (WT in CD45.1). The ratio of CD45.1 to CD45.2 positive cells in the BM, PB, thymus,
and spleen of recipient mice was measured by flow cytometry. IN VIVO HOMING ASSAY For in vivo homing assay64, recipient mice were lethally irradiated (10 Gy) 24 h before LSK
transplantation. Freshly isolated LSK cells from 8-week-old mice were labeled with 1 μM CFSE (Thermo Fisher Scientific C34554) per the manufacturer’s instructions. CFSE-labeled LSK cells (2
× 105) were then resuspended in PBS and intravenously injected into recipient mice. Sixteen hours after injection, mice were sacrificed, and the frequencies of CFSE+ cells in the BM, PB, and
spleen were determined by flow cytometry. To determine the frequency of clonogenic progenitors, mononuclear cells from the BM, PB, and spleen were subjected to CFU assays as described
above. To block the interaction between VLA-4 and VCAM-1, CFSE-labeled LSK cells were incubated with an anti-VCAM-1 Ab (2 mg kg−1) or an isotype control, and the cells were intravenously
injected into recipients 1 h before transplantation. TRANS-STROMAL MIGRATION AND ADHESION ASSAY For the trans-stromal migration assay65, LSK cells (1 × 105 cells chamber−1) were loaded into
the top chamber of a 24-well transwell plate containing a monolayer of BMSCs. Recombinant mouse CXCL12 (100 ng mL−1; R&D Systems P40224) was loaded into the bottom chamber. The plate was
then incubated at 37 °C in 5% CO2 for 16 h. After incubation, the number of migrating LSK cells in the bottom chamber was calculated. To perform the adhesion assay, CFSE-labeled LSK cells
(5 × 104) were co-cultured with BMSCs (2 × 105) in 24-well cell culture plates. Three h after co-culture, nonadherent LSK cells were removed via three gentle washes with PBS, and adherent
LSK cells were harvested. The frequency of CFSE+ LSK cells was determined by flow cytometry. To block the VLA-4 and VCAM-1 interaction, LSK cells were preincubated with anti-VLA-4 (10 μg
mL−1; Merck CBL1304) Ab or an isotype control (10 μg ml−1; BD Biosciences 559478) for 30 min, and BMSCs were preincubated with anti-VCAM-1 Ab (10 μg mL−1) or an isotype control for 30 min.
MRNA-SEQ DATA ANALYSIS Total RNAs from WT and KO BM cells were extracted using TRIzol (Thermo Fisher Scientific 15596026) according to the manufacturer’s protocol. Subsequently, 1 μg of
total RNA was used to construct cDNA libraries using TruSeq RNA library kit (Illumina, RS-122-2001) according to the manufacturer’s protocol. The size distribution and quality of cDNA
libraries were monitored by Agilent 2100 Bioanalyzer (Agilent Technologies) and quantitative PCR using Kapa Library Quantification Kit (Kapa Biosystems KK4824). After the quality check, cDNA
libraries were sequenced using an Illumina HiSeq4000 sequencer (Illumina). After removing low quality and adapter sequences, the raw reads were aligned to the reference genome _Mus
musculus_ (mm10) (genome assembly information access, GCF_000001635.20) using HISAT v2.0.566. After aligning reads to the genome, StringTie v1.3.3b was used to assemble transcripts and
estimate their abundance measured in fragments per kilobase of exon per million fragments mapped (FPKM)67,68. False discovery rate (FDR) was controlled by adjusting _p_-value cutoff of 0.05
using Benjamini–Hochberg algorithm69. Subsequently, hierarchical clustering analysis was performed to analyze differentially expressed gene (DEG) set using complete linkage and Euclidean
distance as a measure of similarity. Gene set enrichment analysis including functional annotation and pathway analyses were performed based on Gene Ontology (www.geneontology.org/) and KEGG
pathway (http://www.genome.jp/kegg/pathway.html). An overview of the gene expression data was deposited at NCBI’s Gene Expression Omnibus (GEO). It is accessible through GEO series accession
number of GSE128705. REAL-TIME QUANTITATIVE RT-PCR ANALYSIS Total RNA was purified from BMSCs using Trizol reagent (Thermo Fisher Scientific 15596026) according to the manufacturer’s
instructions; it was then reverse transcribed into cDNA using a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific K1621). After cDNA synthesis, quantitative PCR (qPCR) was
conducted to measure mRNA levels of mouse _Alcam_, _Angpt1_, _Cdh2_, _Ctsk_, _Cxcl12_, _Fn1_, _Kitl_, _Icam1_, _p16__INK4a_, _p19__ARF_, _Selp_, _Vcam1,_ and _Gapdh_ using a 7500 Real-time
PCR system (Thermo Fisher Scientific). _Gapdh_ was used as an internal control to quantify the levels of other mRNA transcripts. The primer pairs used for qRT-PCR are listed in Supplementary
Table 1. INHIBITION OF _PHC2_ EXPRESSION BY SHRNA Lentiviral plasmids expressing shRNAs directed against several distinct regions of _Phc2_ (SHCLNG-NM_018774) and control shRNA plasmids
(SHC001) were purchased from Sigma-Aldrich. OP9 cells (mouse BMSC line, ATCC CRL-2749) were transfected with each shRNA-containing plasmid using Lipofectamine 2000 transfection reagent
(Thermo Fisher Scientific 11668019) according to the manufacturer’s instructions. After transfection, inhibition of _Phc2_ expression was determined by qRT-PCR and immunoblotting. Primer
sequences for _Phc2_ for qRT-PCR are listed in Supplementary Table 1. CHIP ASSAY The lysates of BM cells (2 × 106) from 8-week-old mice were prepared for the ChIP assay70. BM cell lysates
were sonicated and immunoprecipitated with the anti-Bmi1 (Santa Cruz Biotechnology, Inc. sc-10745, 2 μg sample−1), anti-Cbx7 (Santa Cruz Biotechnology, Inc. sc-70232, 2 μg sample−1),
anti-H2AK119ub (Cell Signaling Technology 8240, 2 μg sample−1), anti-H3K9ac (Merck 06-942, 2 μg sample−1), anti-H3K4me3 (Abcam ab12209, 2 μg sample−1), anti-H3K27me3 (Merck 07-449, 2 μg
sample−1), anti-Phc2 (Santa Cruz Biotechnology, Inc. sc-160664, 2 μg sample−1), and anti-Ring1b (Cell Signaling Technology 5694, 2 μg sample−1) Abs. Additionally, GSK126-treated OP9 cell
lysates were sonicated and immunoprecipitated with the anti-Bmi1, anti-Phc2, and anti-Ring1b Abs. After immunoprecipitation, immune complexes were collected with protein A agarose (Merck
GE17-0963-03) and extracted with an extraction buffer (1% SDS, 0.1 M NaHCO2). DNA cross-links were reversed by heating to 65 °C for 8 h. DNA was extracted with phenol/chloroform and
precipitated with ethanol. DNA isolated from an aliquot of total nuclear extract was used as the loading control for PCR (input control). qPCR was performed as described above. Data are
presented after normalizing each immunoprecipitated DNA Ct value to 10% of the input DNA Ct value. IN VIVO ADMINISTRATION OF GSK126 WT mice were intraperitoneally injected with 50 or 100 mg
kg−1 of DMSO-dissolved GSK126. Mice were sacrificed at 16 h after GSK126 injection and the expression of VCAM-1 from BMSCs of sacrificed mice were analyzed by immunoblotting and flow
cytometry. In addition, trans-stromal migration and adhesion assays using LSK cells and BMSCs of sacrificed mice were performed as described above. STATISTICAL ANALYSIS Mean values among
more than three experimental groups were compared by one-way ANOVA with Tukey HSD analysis and comparison between two groups was analyzed by two-tailed Student’s _t_-test using SPSS
statistics software, ver. 24.0 (SPSS Inc). All data are presented as the mean ± standard error of mean (SEM). In each graph, significant differences as determined by _p_-values less than
0.05 and 0.01 are indicated by asterisks (* and **, respectively). REPORTING SUMMARY Further information on research design is available in the Nature Research Reporting Summary linked to
this article. DATA AVAILABILITY The raw mRNA-seq data were deposited into the Sequence Read Archive (SRA) of National Center for Biotechnology Information (NCBI) with the following accession
numbers: GSM3683312, GSM3683313, GSM3683314, GSM3683315, GSM3683316, and GSM3683317. An overview of the gene expression data was deposited at NCBI’s Gene Expression Omnibus (GEO). It is
accessible through GEO series accession number of GSE128705. Source data underlying Figs. 1–8, Tables 1–2, Supplementary Figs. 1–7, and Supplementary Figs. 10–18 are provided as a Source
Data file. All other data that support the findings of this study are available from the corresponding author upon reasonable request. REFERENCES * Wright, D. E., Wagers, A. J., Gulati, A.
P., Johnson, F. L. & Weissman, I. L. Physiological migration of hematopoietic stem and progenitor cells. _Science_ 294, 1933–1936 (2001). Article ADS CAS Google Scholar * Laird, D.
J., von Andrian, U. H. & Wagers, A. J. Stem cell trafficking in tissue development, growth, and disease. _Cell_ 132, 612–630 (2008). Article CAS Google Scholar * Bhattacharya, D.,
Rossi, D. J., Bryder, D. & Weissman, I. L. Purified hematopoietic stem cell engraftment of rare niches corrects severe lymphoid deficiencies without host conditioning. _J. Exp. Med._
203, 73–85 (2006). Article CAS Google Scholar * Morrison, S. J. & Spradling, A. C. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. _Cell_ 132,
598–611 (2008). Article CAS Google Scholar * Mazo, I. B., Massberg, S. & von Andrian, U. H. Hematopoietic stem and progenitor cell trafficking. _Trends Immunol._ 32, 493–503 (2011).
Article CAS Google Scholar * Wilson, A. & Trumpp, A. Bone-marrow haematopoietic-stem-cell niches. _Nat. Rev. Immunol._ 6, 93–106 (2006). Article CAS Google Scholar * Mercier, F.
E., Ragu, C. & Scadden, D. T. The bone marrow at the crossroads of blood and immunity. _Nat. Rev. Immunol._ 12, 49–60 (2012). Article CAS Google Scholar * Piunti, A. &
Shilatifard, A. Epigenetic balance of gene expression by Polycomb and COMPASS families. _Science_ 352, aad9780 (2016). Article Google Scholar * Cao, R. et al. Role of histone H3 lysine 27
methylation in Polycomb-group silencing. _Science_ 298, 1039–1043 (2002). Article ADS CAS Google Scholar * Gao, Z. et al. PCGF homologs, CBX proteins, and RYBP define functionally
distinct PRC1 family complexes. _Mol. Cell._ 45, 344–356 (2012). Article CAS Google Scholar * Morey, L., Aloia, L., Cozzuto, L., Benitah, S. A. & Di Croce, L. RYBP and Cbx7 define
specific biological functions of polycomb complexes in mouse embryonic stem cells. _Cell Rep._ 3, 60–69 (2013). Article CAS Google Scholar * Wang, H. et al. Role of histone H2A
ubiquitination in Polycomb silencing. _Nature_ 431, 873–878 (2004). Article ADS CAS Google Scholar * Tavares, L. et al. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target
sites independently of PRC2 and H3K27me3. _Cell_ 148, 664–678 (2012). Article CAS Google Scholar * Isono, K. et al. Mammalian polyhomeotic homologues Phc2 and Phc1 act in synergy to
mediate polycomb repression of Hox genes. _Mol. Cell. Biol._ 25, 6694–6706 (2005). Article CAS Google Scholar * Petit, I. et al. G-CSF induces stem cell mobilization by decreasing bone
marrow SDF-1 and up-regulating CXCR4. _Nat. Immunol._ 3, 687–694 (2002). Article CAS Google Scholar * Bendall, L. J. & Bradstock, K. F. G-CSF: from granulopoietic stimulant to bone
marrow stem cell mobilizing agent. _Cytokine Growth Factor Rev._ 25, 355–367 (2014). Article CAS Google Scholar * Broxmeyer, H. E. et al. Rapid mobilization of murine and human
hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. _J. Exp. Med._ 201, 1307–1318 (2005). Article CAS Google Scholar * Devine, S. M. et al. Rapid mobilization of
functional donor hematopoietic cells without G-CSF using AMD3100, an antagonist of the CXCR4/SDF-1 interaction. _Blood_ 112, 990–998 (2008). Article CAS Google Scholar * Heissig, B. et
al. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. _Cell_ 109, 625–637 (2002). Article CAS Google Scholar * Aicher, A.
et al. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. _Nat. Med._ 9, 1370–1376 (2003). Article CAS Google Scholar * Calvi, L. et al.
Osteoblastic cells regulate the haematopoietic stem cell niche. _Nature_ 425, 841–846 (2003). Article ADS CAS Google Scholar * Kiel, M. J. et al. SLAM family receptors distinguish
hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. _Cell_ 121, 1109–1121 (2005). Article CAS Google Scholar * Méndez-Ferrer, S. et al. Mesenchymal and
haematopoietic stem cells form a unique bone marrow niche. _Nature_ 466, 829–834 (2010). Article ADS Google Scholar * Iademarco, M. F., Barks, J. L. & Dean, D. C. Regulation of
vascular cell adhesion molecule-1 expression by IL-4 and TNF-alpha in cultured endothelial cells. _J. Clin. Invest._ 95, 264–271 (1995). Article CAS Google Scholar * McCabe, M. T. et al.
EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. _Nature_ 492, 108–112 (2012). Article ADS CAS Google Scholar * Koni, P. A. et al. Conditional
vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. _J. Exp. Med._ 193, 741–754 (2001). Article CAS Google Scholar * Ulyanova, T. et al.
VCAM-1 expression in adult hematopoietic and nonhematopoietic cells is controlled by tissue-inductive signals and reflects their developmental origin. _Blood_ 106, 86–94 (2005). Article CAS
Google Scholar * Scott, L. M., Priestley, G. V. & Papayannopoulou, T. Deletion of α4 integrins from adult hematopoietic cells reveals roles in homeostasis, regeneration, and homing.
_Mol. Cell. Biol._ 23, 9349–9360 (2003). Article CAS Google Scholar * Priestley, G. V., Ulyanova, T. & Papayannopoulou, T. Sustained alterations in biodistribution of stem/progenitor
cells in Tie2Cre+ α4f/f mice are hematopoietic cell autonomous. _Blood_ 109, 109–111 (2007). Article CAS Google Scholar * Papayannopoulou, T., Priestley, G. V., Nakamoto, B.,
Zafiropoulos, V. & Scott, L. M. Molecular pathways in bone marrow homing: dominant role of α4β1 over β2-integrins and selectins. _Blood_ 98, 2403–2411 (2001). Article CAS Google
Scholar * Papayannopoulou, T. & Nakamoto, B. Peripheralization of hemopoietic progenitors in primates treated with anti-VLA4 integrin. _Proc. Natl Acad. Sci. USA_ 90, 9374–9378 (1993).
Article ADS CAS Google Scholar * Kavanagh, D. P. et al. Haematopoietic stem cell recruitment to injured murine liver sinusoids depends on α4β1 integrin/VCAM-1 interactions. _Gut_ 59,
79–87 (2010). Article CAS Google Scholar * Osborn, L. et al. Direct expression cloning of vascular cell adhesion molecule 1, a cytokine-induced endothelial protein that binds to
lymphocytes. _Cell_ 59, 1203–1211 (1989). Article CAS Google Scholar * Miyake, K. et al. A VCAM-like adhesion molecule on murine bone marrow stromal cells mediates binding of lymphocyte
precursors in culture. _J. Cell Biol._ 114, 557–565 (1991). Article CAS Google Scholar * Jacobsen, K., Kravitz, J., Kincade, P. & Osmond, D. Adhesion receptors on bone marrow stromal
cells: in vivo expression of vascular cell adhesion molecule-1 by reticular cells and sinusoidal endothelium in normal and gamma-irradiated mice. _Blood_ 87, 73–82 (1996). CAS PubMed
Google Scholar * Schweitzer, K. M. et al. Constitutive expression of E-selectin and vascular cell adhesion molecule-1 on endothelial cells of hematopoietic tissues. _Am. J. Pathol._ 148,
165–175 (1996). CAS PubMed PubMed Central Google Scholar * Freedman, A. S. et al. Adhesion of human B cells to germinal centers in vitro involves VLA-4 and INCAM-110. _Science_ 249,
1030–1033 (1990). Article ADS CAS Google Scholar * Schlesinger, M. & Bendas, G. Vascular cell adhesion molecule‐1 (VCAM‐1)—an increasing insight into its role in tumorigenicity and
metastasis. _Int. J. Cancer_ 136, 2504–2514 (2015). Article CAS Google Scholar * Bradstock, K. F. & Gottlieb, D. J. Interaction of acute leukemia cells with the bone marrow
microenvironment: implications for control of minimal residual disease. _Leuk. Lymphoma_ 18, 1–16 (1995). Article CAS Google Scholar * Winter, S. S. et al. Enhanced T‐lineage acute
lymphoblastic leukaemia cell survival on bone marrow stroma requires involvement of LFA‐1 and ICAM‐1. _Br. J. Haematol._ 115, 862–871 (2001). Article CAS Google Scholar * Damiano, J. S.,
Cress, A. E., Hazlehurst, L. A., Shtil, A. A. & Dalton, W. S. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines.
_Blood_ 93, 1658–1667 (1999). CAS PubMed PubMed Central Google Scholar * Tabe, Y. et al. Activation of integrin-linked kinase is a critical prosurvival pathway induced in leukemic cells
by bone marrow-derived stromal cells. _Cancer Res._ 67, 684–694 (2007). Article CAS Google Scholar * Jacamo, R. et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of
NF-κB mediates chemoresistance. _Blood_ 123, 2691–2702 (2014). Article CAS Google Scholar * Cullen, S. M., Mayle, A., Rossi, L. & Goodell, M. A. Hematopoietic stem cell development:
an epigenetic journey. _Curr. Top. Dev. Biol._ 107, 39–75 (2014). Article CAS Google Scholar * Hu, D. & Shilatifard, A. Epigenetics of hematopoiesis and hematological malignancies.
_Genes Dev._ 30, 2021–2041 (2016). Article CAS Google Scholar * Takamatsu-Ichihara, E. & Kitabayashi, I. The roles of Polycomb group proteins in hematopoietic stem cells and
hematological malignancies. _Int. J. Hematol._ 103, 634–642 (2016). Article CAS Google Scholar * Vidal, M. & Starowicz, K. Polycomb complexes PRC1 and their function in hematopoiesis.
_Exp. Hematol._ 48, 12–31 (2017). Article CAS Google Scholar * Sato, T. et al. Mammalian Polycomb complexes are required for Peyer’s patch development by regulating lymphoid cell
proliferation. _Gene_ 379, 166–174 (2006). Article CAS Google Scholar * Ohta, H. et al. Polycomb group gene rae28 is required for sustaining activity of hematopoietic stem cells. _J. Exp.
Med._ 195, 759–770 (2002). Article CAS Google Scholar * Kim, J. Y. et al. Defective long-term repopulating ability in hematopoietic stem cells lacking the polycomb-group gene rae28.
_Eur. J. Haematol._ 73, 75–84 (2004). Article CAS Google Scholar * Hidalgo, I. et al. Ezh1 is required for hematopoietic stem cell maintenance and prevents senescence-like cell cycle
arrest. _Cell Stem Cell_ 11, 649–662 (2012). Article CAS Google Scholar * Xie, H. et al. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a
developmental-stage-specific manner. _Cell Stem Cell_ 14, 68–80 (2014). Article CAS Google Scholar * van Den Boom, V. et al. Nonredundant and locus-specific gene repression functions of
PRC1 paralog family members in human hematopoietic stem/progeitor cells. _Blood_ 121, 2452–2461 (2013). Article Google Scholar * Klauke, K. et al. Polycomb Cbx family members mediate the
balance between haematopoietic stem cell self-renewal and differentiation. _Nat. Cell Biol._ 15, 353–362 (2013). Article CAS Google Scholar * Gray, F. et al. BMI1 regulates PRC1
architecture and activity through homo-and hetero-oligomerization. _Nat. Commun._ 7, 13343 (2016). Article ADS CAS Google Scholar * Peister, A. et al. Adult stem cells from bone marrow
(MSCs) isolated from different strains of inbred mice vary in surface epitopes, rates of proliferation, and differentiation potential. _Blood_ 103, 1662–1668 (2004). Article CAS Google
Scholar * Broske, A. M. et al. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. _Nat. Genet._ 41, 1207–1215 (2009). Article Google Scholar *
Vukovic, M. et al. Adult hematopoietic stem cells lacking Hif-1α self-renew normally. _Blood_ 127, 2841–2846 (2016). Article CAS Google Scholar * Han, J. et al. Autophagy induced by AXL
receptor tyrosine kinase alleviates acute liver injury via inhibition of NLRP3 inflammasome activation in mice. _Autophagy_ 12, 2326–2343 (2016). Article CAS Google Scholar * Goldman, D.
C. et al. BMP4 regulates the hematopoietic stem cell niche. _Blood_ 114, 4393–4401 (2009). Article CAS Google Scholar * Ramirez, P. et al. BIO5192, a small molecule inhibitor of VLA-4,
mobilizes hematopoietic stem and progenitor cells. _Blood_ 114, 1340–1343 (2009). Article CAS Google Scholar * Chen., Z. et al. Wip1 deficiency impairs haematopoietic stem cell function
via p53 and mTORC1 pathways. _Nat. Commun._ 6, 6808 (2015). Article ADS CAS Google Scholar * Papayannopoulou, T., Priestley, G. V. & Nakamoto, B. Anti-VLA4/VCAM-1-induced
mobilization requires cooperative signaling through the kit/mkit ligand pathway. _Blood_ 91, 2231–2239 (1998). CAS PubMed Google Scholar * Foudi, A. et al. Reduced retention of
radioprotective hematopoietic cells within the bone marrow microenvironment in _CXCR4_ –/– chimeric mice. _Blood_ 107, 2243–2251 (2006). Article CAS Google Scholar * Gonzalez-Nieto, D. et
al. Connexin-43 in the osteogenic BM niche regulates its cellular composition and the bidirectional traffic of hematopoietic stem cells and progenitors. _Blood_ 119, 5144–5154 (2012).
Article CAS Google Scholar * Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. _Nat. Methods_ 12, 357–360 (2015). Article CAS
Google Scholar * Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. _Nat. Biotechnol._ 33, 290–295 (2015). Article CAS Google Scholar *
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. _Nat. Protoc._ 11,
1650–1667 (2016). Article CAS Google Scholar * Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. _J. R. Soc.
Ser. B Stat. Methodol._ 57, 289–300 (1995). MathSciNet MATH Google Scholar * Cho, K. W., Bae, J., Lee, S. J. & Chun, T. Expression pattern and functional role of Phc2 during
activation of helper T cells after antigenic stimulation. _In Vitro Cell Dev. Biol. Anim_ 49, 360–370 (2013). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank
Amgen Inc. (Thousand Oaks, CA, USA) for providing G-CSF. We also thank Eun-Kyeong Jo (Chungnam National University School of Medicine), Kyung-Mi Lee (Korea University College of Medicine),
Seungkwon You (Korea University School of Life Sciences and Biotechnology), and Seok-Ho Hong (Kangwon National University College of Medicine) for their critical reading of our manuscript or
helpful suggestions. This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Ministry of Science &
ICT (2017M3A9C8060392), and an intramural research program funded by Korea University grant (2016). AUTHOR INFORMATION Author notes * These authors contributed equally: Joonbeom Bae,
Sang-Pil Choi. AUTHORS AND AFFILIATIONS * Department of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul, 02841, Republic of Korea Joonbeom Bae, Sang-Pil
Choi, Si-Won Park, Chang-Yong Choi, Jihye Han, Sang-Hoon Kim, Han-Hyoung Lee, Kyungmin Park, Young Sik Lee & Taehoon Chun * Laboratory Animal Center, Wakayama Medical University,
Wakayama, 641-8509, Japan Kyoichi Isono * Department of Biomedical Science, CHA University, Seongnam, Gyounggi-do, 13488, Republic of Korea Ji Yoon Lee * Department of Urology, School of
Medicine, University of California, San Francisco, San Francisco, CA, 94158, USA Hyun Yong Jin * Department of Biomedical Laboratory Science, College of Health Science, Cheongju University,
Cheongju-si, 28503, Republic of Korea Suk Jun Lee * Department of Microbiology and Immunology, Cancer Research Institute and Xenotransplantation Research Center, Seoul National University
College of Medicine, Seoul, 03087, Republic of Korea Chung-Gyu Park * Laboratory for Developmental Genetics, RIKEN Center for Integrative Medical Sciences, Yokohama, 230-0045, Japan Haruhiko
Koseki Authors * Joonbeom Bae View author publications You can also search for this author inPubMed Google Scholar * Sang-Pil Choi View author publications You can also search for this
author inPubMed Google Scholar * Kyoichi Isono View author publications You can also search for this author inPubMed Google Scholar * Ji Yoon Lee View author publications You can also search
for this author inPubMed Google Scholar * Si-Won Park View author publications You can also search for this author inPubMed Google Scholar * Chang-Yong Choi View author publications You can
also search for this author inPubMed Google Scholar * Jihye Han View author publications You can also search for this author inPubMed Google Scholar * Sang-Hoon Kim View author publications
You can also search for this author inPubMed Google Scholar * Han-Hyoung Lee View author publications You can also search for this author inPubMed Google Scholar * Kyungmin Park View author
publications You can also search for this author inPubMed Google Scholar * Hyun Yong Jin View author publications You can also search for this author inPubMed Google Scholar * Suk Jun Lee
View author publications You can also search for this author inPubMed Google Scholar * Chung-Gyu Park View author publications You can also search for this author inPubMed Google Scholar *
Haruhiko Koseki View author publications You can also search for this author inPubMed Google Scholar * Young Sik Lee View author publications You can also search for this author inPubMed
Google Scholar * Taehoon Chun View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS T.C. conceived, designed, and supervised this study. J.B.,
S.-P.C., K.I., J.Y.L., S.-W.P., C.-Y.C., J.H., S.-H.K., H.-H.L., K.P. and S.J.L. performed experiments. J.B., S.-P.C., C.-Y.C., C.-G.P. and T.C. analyzed and interpreted data. J.B., S.-P.C.,
H.Y.J., C.-G.P., H.K., Y.S.L. and T.C. discussed, wrote and edited the manuscript. All authors reviewed and approved the manuscript. CORRESPONDING AUTHOR Correspondence to Taehoon Chun.
ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PEER REVIEW INFORMATION: _Nature Communications_ thanks Hal Broxmeyer and other
anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. PUBLISHER’S NOTE: Springer Nature remains neutral with regard to
jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS
AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in
any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The
images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly
from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Bae, J., Choi,
SP., Isono, K. _et al._ Phc2 controls hematopoietic stem and progenitor cell mobilization from bone marrow by repressing _Vcam1_ expression. _Nat Commun_ 10, 3496 (2019).
https://doi.org/10.1038/s41467-019-11386-4 Download citation * Received: 18 April 2018 * Accepted: 12 July 2019 * Published: 02 August 2019 * DOI: https://doi.org/10.1038/s41467-019-11386-4
SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to
clipboard Provided by the Springer Nature SharedIt content-sharing initiative