Multiplexed relative and absolute quantitative immunopeptidomics reveals mhc i repertoire alterations induced by cdk4/6 inhibition
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Peptides bound to class I major histocompatibility complexes (MHC) play a critical role in immune cell recognition and can trigger an antitumor immune response in cancer. Surface
MHC levels can be modulated by anticancer agents, altering immunity. However, understanding the peptide repertoire’s response to treatment remains challenging and is limited by quantitative
mass spectrometry-based strategies lacking normalization controls. We describe an experimental platform that leverages recombinant heavy isotope-coded peptide MHCs (hipMHCs) and multiplex
isotope tagging to quantify peptide repertoire alterations using low sample input. HipMHCs improve quantitative accuracy of peptide repertoire changes by normalizing for variation across
analyses and enable absolute quantification using internal calibrants to determine copies per cell of MHC antigens, which can inform immunotherapy design. Applying this platform in melanoma
cell lines to profile the immunopeptidome response to CDK4/6 inhibition and interferon-γ — known modulators of antigen presentation — uncovers treatment-specific alterations, connecting the
intracellular response to extracellular immune presentation. SIMILAR CONTENT BEING VIEWED BY OTHERS VALIDATION AND QUANTIFICATION OF PEPTIDE ANTIGENS PRESENTED ON MHCS USING SUREQUANT
Article 22 October 2024 SINGLE-CELL DERIVED TUMOR ORGANOIDS DISPLAY DIVERSITY IN HLA CLASS I PEPTIDE PRESENTATION Article Open access 21 October 2020 CHARACTERIZATION OF AN EXPANDED SET OF
ASSAYS FOR IMMUNOMODULATORY PROTEINS USING TARGETED MASS SPECTROMETRY Article Open access 25 June 2024 INTRODUCTION Cells present signals on the extracellular surface that serve as targets
for immune cell recognition. These signals, peptides presented by class I major histocompatibility complexes (MHCs), are typically derived from intracellular source proteins, and may
therefore provide an external representation of the internal cell state1. As a reflection of this, the peptide MHC (pMHC) repertoire, or “immunopeptidome”, of cancer cells may contain
tumor-associated or mutation-containing antigens that serve as tumor-specific markers to activate T cells and initiate an antitumor immune response. This interaction can be strengthened with
checkpoint blockade (CB) immunotherapies; however, low response rates and toxicity remain barriers to their broad clinical success2,3. A growing body of evidence suggests that combining CB
with other treatments, such as small-molecule inhibitors, cytotoxic agents, and radiotherapy, could potentiate the response to CB, in part, by augmenting tumor immunogenicity through
increased surface pMHC expression4,5,6,7. While clinical trials in this space have shown promise8,9, the optimal combination of agents, as well as the order and timing of administration, are
only beginning to be understood. In order to improve combinatorial strategies, a quantitative, molecular understanding of how different perturbations shift the immunopeptidome is required.
Furthermore, achieving absolute quantification of presented antigens is necessary to inform immunotherapy drug design, as targeted strategies have varying thresholds of antigen expression
required for an optimal antitumor response. Traditional data-dependent acquisition (DDA) methods to profile pMHC repertoires using mass spectrometry (MS) are well documented10,11,12, but
quantitative methods have critical limitations. Specifically, most common relative quantification pMHC methods lack a normalization strategy to account for variations in sample input and
processing13,14,15,16,17,18. Peptide losses during processing vary across peptide sequences, concentrations, and samples, underscoring the need for normalization19,20. Absolute
quantification of pMHCs to date is most commonly performed by comparing endogenous levels of pMHCs to exogenous peptide standards, again failing to account for sample losses21,22,23,24.
Losses can be accounted for with internal pMHC standards, but require laborious refolding of pMHCs for every target of interest19,25. Nevertheless, this approach relies on single point
calibration, ignoring the effects of ion suppression, thereby inaccurately estimating absolute pMHC levels in quantitative analyses. To combat these challenges in quantitative
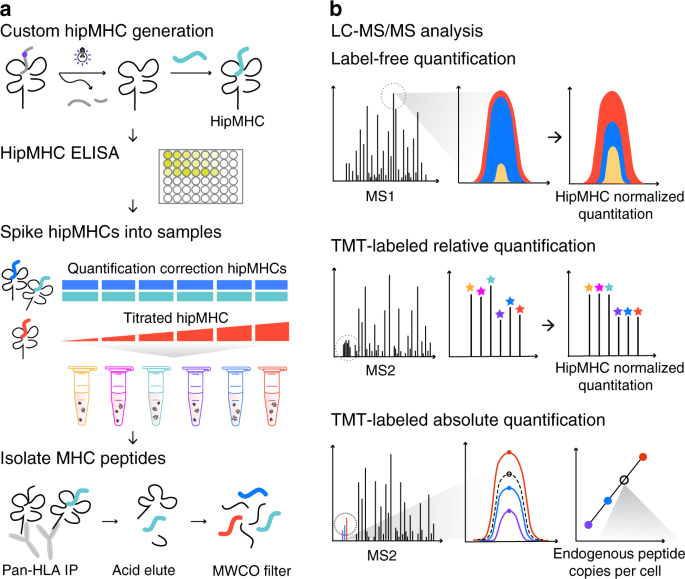
immunopeptidomic profiling, we present a platform that utilizes ultraviolet (UV)-mediated peptide exchange of recombinant MHC monomers to generate on demand heavy-isotope-labeled pMHCs for
relative and absolute quantification of pMHC repertoires using low sample input. We demonstrate that the addition of heavy-isotope pMHCs (hipMHCs) spiked into sample lysates for
normalization improves quantitative accuracy between samples for both label-free (LF) and multiplexed (tandem mass tags (TMTs) labeled) analyses and provides an estimate of ion suppression
through regression against a titrated internal calibrant. Furthermore, we utilize hipMHC multipoint-embedded standard curves coupled with isobaric mass tags to accurately quantify the
absolute number of copies per cell of target antigens within a single analysis. We apply this platform to profile immunopeptidomic changes in melanoma cell lines, comparing treatment with
palbociclib (a small-molecule CDK4/6 inhibitor) and interferon-γ (IFN-γ), both known modulators of antigen presentation7,26. Peptides derived from proteins implicated in the biological
response to palbociclib and IFN-γ are selectively enriched in the pMHC repertoire following treatment, connecting the intracellular response to extracellular immune presentation.
Furthermore, peptides derived from the metabolic response to palbociclib, along with known tumor-associated antigens (TAAs), display significantly increased presentation with palbociclib
treatment. We propose this platform can be broadly applied to profile immunopeptidomic changes in a high-throughput, low-input format across sample types and treatments to inform combination
therapy strategies and can be used to identify and quantify treatment-modulated antigen targets for targeted immunotherapy. RESULTS PLATFORM FOR RELATIVE AND ABSOLUTE PMHC QUANTITATION We
set out to develop a platform to provide accurate relative and absolute quantification of pMHCs across multiple samples while controlling for losses associated with sample processing and
enrichment. Accurate quantitative analysis is best performed with internal standards and multipoint internal calibration curves. To generate internal standards, heavy-isotope-labeled MHC
peptides of interest were synthesized and loaded onto biotinylated MHC monomers through UV-mediated peptide exchange27 (Fig. 1a). To control for loading efficiency of synthetic peptides into
recombinant MHC proteins, the concentration of stable hipMHC complexes was determined by an enzyme-linked immunosorbent assay (ELISA). Stable hipMHC complexes were then used in two ways:
selected hipMHC complexes were spiked at the same concentration into the whole-cell lysate from each sample to provide a normalization correction for relative quantification across samples,
while other hipMHC complexes were titrated at different concentrations into each sample to verify correction parameters, estimate dynamic range suppression for quantification, and/or create
an internal standard curve for absolute quantification of a specific peptide. After adding hipMHCs, endogenous and exogenous pMHCs were isolated by immunoprecipitation (IP), acid elution,
and molecular weight size-exclusion filtration. Peptide mixtures were next analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) in three different ways (Fig. 1b). For LF
analyses, samples were analyzed individually, and peptides were quantified by integrating the area under the curve (AUC) for the chromatographic elution of precursor masses for each
peptide-spectrum match (PSM). Relative AUC intensities of quantification correction hipMHCs were used to normalize AUC intensities of endogenous peptides across analyses. To analyze multiple
samples simultaneously, we labeled samples with TMT and relative TMT ion intensity ratios of hipMHCs were used for normalization to correct the relative quantification in multiplexed
samples. TMT-labeled titrated hipMHCs were also used for absolute quantification of endogenous peptides. Apex TMT intensities of hipMHCs generated a peptide-specific multipoint calibration
curve to calculate the average number of copies per cell. As a control, heavy-isotope-coded synthetic peptides not complexed to MHCs were spiked into the whole-cell lysate prior to IP. These
peptides were not detected in the subsequent LC-MS/MS analysis, demonstrating that only peptides in stable complexes were isolated in our workflow and that excess free peptides did not
displace endogenously presented peptides. HIPMHC STANDARDS IMPROVE QUANTITATIVE ACCURACY To demonstrate the improved quantitative accuracy obtained with hipMHCs, we used five LF and six
TMT-labeled technical replicates of 1 × 107 MDA-MB-231 breast cancer cells to measure variance between replicates before and after hipMHC correction (Fig. 2a, Supplementary Data 1). In both
LF and TMT-labeled workflows, we spiked 30 fmol of two quantification correction hipMHCs into each sample, as adding additional correction hipMHCs gave minimal improvement in quantitative
accuracy. We also added 30–300 fmol of a titrated hipMHC across samples (Fig. 2b). A total of 2369 unique pMHCs were identified in total across five LF analyses, 1352 of which were
quantifiable via AUC integration (Fig. 2c). Of these quantifiable peptides, only 589 were quantified in all five analyses, highlighting the poor run-to-run overlap of LF analyses, even with
replicate samples (Supplementary Fig. 1a). By comparison, 1754 unique peptides were quantifiable with TMT-labeled analyses. The extra sample handling steps associated with TMT labeling can
result in losses, so to achieve high coverage of the immunopeptidome, labeled samples were divided into six separate analyses, thereby increasing the number of unique identifications
(Supplementary Fig. 1b). In both LF and TMT analyses, peptides matched expected length distributions of 8–11 amino acids (Supplementary Fig. 1c), and 82% of LF and 92% of TMT 9-mers were
predicted to be binders with <500 nM predicted affinity28 (Fig. 2d). The peptides were similarly apportioned across alleles between LF and TMT analyses, and the allele motifs aligned with
those previously reported29 (Supplementary Fig. 1d, e). Further reducing the input material to 5 × 106 cells still resulted in 86% of the number of unique peptides identified with 1 × 107
cells in a single LF analysis, establishing the sensitivity of this method for low-input pMHC analyses (Supplementary Fig. 1f). To normalize LF and TMT-labeled data sets, we applied
correction parameters calculated from the quantification correction hipMHCs. The titrated peptide, SVVESVKFL7, displayed an improved linear fit after correction, with an even more pronounced
effect in the LF samples (Fig. 2e). We observed dynamic range suppression for this peptide in TMT-labeled (4.7×) and LF (2.7×) data sets, demonstrating in both cases that quantitative
differences are likely larger than what is measured. In both analyses, hipMHC quantification correction reduced variation, for example, peptides from TMT-labeled sample 5 have lower
intensities than the other samples, which was corrected by hipMHC normalization (Fig. 2f). The standard deviation from the mean for replicate samples decreased with hipMHC correction in LF
and TMT-labeled samples (Fig. 2g), although TMT labeling showed lower variation between replicates (Fig. 2h), allowing for higher confidence in small shifts within the immunopeptidome. We
investigated whether peptides with lower abundance had higher quantitative variation across samples, but found no correlation in LF or TMT-labeled analyses (Supplementary Fig. 1g). ABSOLUTE
QUANTIFICATION OF ENDOGENOUS PMHCS To demonstrate the ability of hipMHCs to quantify pMHC copies per cell, we selected two peptides identified in TMT-labeled MDA-MB-231 cells for absolute
quantification: KLDVGNAEV derived from B cell receptor-associated protein 31 (BCAP31) and KQVSDLISV from DEAD-box RNA helicase 5 (DDX5). BCAP31 regulates the transport of membrane proteins
from the endoplasmic reticulum to the Golgi, a central component of antigen processing, and is a known TAA peptide30. DDX5 is important in gene expression regulation and has been implicated
in proliferation, metastasis, and tumorigenesis in cancer31. These peptides were detected at differing levels with the highly abundant BCAP31 peptide falling in the 98th percentile and DDX5
falling in the 33rd percentile of abundance (Fig. 3a). Both peptides were synthesized with heavy-isotope-labeled leucine (+7), and hipMHC normalization standards were added to three
replicates of 1 × 107 MDA-MB-231 cells along with titrated amounts of BCAP31 and DDX5 hipMHC (Fig. 3b). We then labeled samples with TMT and performed LC-MS/MS analysis using an inclusion
list, so only targeted peptides of interest were selected for fragmentation (Supplementary Data 2). Chromatographic traces of the three TMT reporter ions for heavy BCAP31 and DDX5 peptides
displayed increasing ion intensities with increasing amount of hipMHC added (Fig. 3c). In order to quantify peptide expression, the apex intensities of reporter ions were adjusted based on
the normalization hipMHCs, and a linear fit was used to determine pMHC concentration present in the sample. Cells had an average of 1740 copies per cell of the BCAP31 peptide, and 81 copies
per cell of the DDX5 peptide (Fig. 3d). Concentrations of the DDX5 hipMHC as low as 100 attomole were detected (six copies per cell) showcasing the broad range of pMHC expression levels
quantifiable by our method (Supplementary Fig. 2). Furthermore, BCAP31 and DDX5 had a dynamic range suppression of 1.9× and 2.5×, respectively, illustrating that the ion suppression is not
uniform across peptides and that peptide-specific internal standards may be required for absolute quantification of each pMHC of interest. CDK4/6 INHIBITION ALTERS THE PMHC REPERTOIRE IN
MELANOMA Cyclin-dependent kinases 4 and 6 (CDK4/6) control cell cycle progression by phosphorylating Rb1, thereby releasing the E2F family of transcription factors that drive progression
through the G1 checkpoint32. CDK4/6 is often dysregulated and overactive in cancer, leading to uncontrolled proliferation33. As such, CDK4/6 inhibitors have emerged as a potentially powerful
class of anticancer agents, active against a spectrum of tumor types including melanoma34. In recent years, CDK4/6 inhibitors have also been shown to enhance tumor immunogenicity by
increasing surface MHC class I expression and boosting T cell activation and infiltration7,35. These data highlight CDK4/6 inhibitors as an attractive candidate to combine with CB or other
immunotherapies to augment immunotherapy response rates in melanoma. However, to date, the effect of CDK4/6 inhibition on the MHC class I peptide repertoire has not been characterized. We
therefore applied our platform to quantify how pMHC repertoires in melanoma change in vitro upon treatment with the CDK4/6 inhibitor, palbociclib, to better understand how CDK4/6 inhibitors
could be leveraged in combination therapy regimes to improve patient outcomes. We selected four melanoma cell lines for analysis: SKMEL5 and SKMEL28 (BRAF mutant), and SKMEL2 and IPC298
(NRAS mutant). Based on sensitivity analyses for each cell line (Fig. 4a), we selected two doses of palbociclib for further study: a low dose of 1 μM, below the half-maximal inhibitory
concentrations (IC50) of all four cell lines, and a high dose of 10 μM, near the IC50. Three biological replicates of 1 × 107 cells of each cell line were then treated with dimethyl
sulfoxide (DMSO), and low- or high-dose palbociclib for 72 h (Fig. 4b). Low-dose treatment increased surface class I MHC presentation, as measured by flow cytometry, by 1.5–2× across cell
lines, whereas high-dose treatment had a milder effect (Fig. 4c, Supplementary Fig. 3a). To characterize the pMHC repertoire alterations induced by palbociclib, multiplexed relative
quantitation was performed comparing low- and high-dose palbociclib to DMSO for each cell line, and data were normalized using hipMHC standards (Supplementary Fig. 3b, Supplementary Data 3).
As with our previous analysis, identified peptides matched expected length distributions, and a majority were predicted to be MHC class I binders (Supplementary Fig. 3c, d).
Immunopeptidomic analysis for each cell line and treatment showed a similar trend to the flow cytometry data: low-dose palbociclib shifted mean pMHC expression higher than DMSO treatment in
all cell lines, and a high-dose palbociclib showed a small increase in mean expression for SKMEL5 compared to DMSO and no significant change for the other cell lines (Fig. 4d, Supplementary
Fig. 3e). We measured a wider distribution of changes in peptide presentation following low-dose treatment, with several peptides increasing eight- to ten-fold, even before considering the
effect of dynamic range suppression (Supplementary Fig. 3b). To gain insight into the biology underlying palbociclib-modulated pMHC alterations, we analyzed our data in two ways. First, we
determined which peptides and source proteins were significantly increased with palbociclib treatment over DMSO. Because many peptides were significantly increased with low-dose treatment,
we also identified the peptides and source proteins that were significantly enriched in presentation with treatment relative to the mean fold change of all peptides, highlighting peptides
preferentially modulated by palbociclib. Using these data, we performed GO term enrichment on the 127 peptides significantly enriched in low-dose-treated SKMEL5 cells (Fig. 4e), and
identified enriched biological processes of interest, including ribosomal biogenesis, glucose metabolic process, and antigen processing, a reflection of the expected biological response to
palbociclib7,36,37 (Fig. 4f, Supplementary Fig. 3f). We performed the same analysis with the raw, non-normalized values, and found only 66 of these peptides were significantly enriched
without hipMHC quantification correction, altering the Gene Oncology (GO) term pathway analysis results (Supplementary Fig. 3g). While peptides mapping to ribosomal biogenesis were still
significantly enriched, the other two biological processes were not, underscoring the importance of using hipMHCs for quantification correction to accurately interpret alterations in pMHC
repertoires. To determine if the measured pMHC alterations to SKMEL5 cells were common across cell lines, we compared the source proteins of peptides that were significantly enriched with
low-dose treatment compared to DMSO across all four cell lines. Surprisingly, a majority (72–88%) of enriched source proteins were unique to each cell line, and we discovered only three
proteins in common: vimentin, putative β-actin-like protein 3, and SIL1 nucleotide exchange factor (Fig. 4g, Supplementary Fig. 3h). Even when comparing source proteins of all peptides
significantly increasing to any extent, just 17 proteins in common were identified, further illustrating the uniqueness of the proteins altered by palbociclib in each immunopeptidomic
landscape (Supplementary Fig. 3i). We investigated whether the commonality of these 17 proteins could be explained by having high abundance in the peptide mixtures, but in SKMEL5 cells they
were scattered throughout the distribution of AUC intensities (Supplementary Fig. 3j). While the list of shared pMHCs and source proteins in common is limited, of interest is the
serine-phosphorylated IRS2 (pIRS2) peptide, RVA[pS]PTSGVK. This post-translationally modified sequence has previously been shown to be restricted to malignant cells, with only the
phosphorylated form demonstrating immunogenic potential38,39. Even though there are no alleles in common across the four cell lines40 (Supplementary Fig. 3k), we observed the pIRS2 peptide
increasing across all cell lines with low-dose treatment (Fig. 4h). Furthermore, RVA[pS]PTSGVK has high expression among pMHCs (Supplementary Fig. 3j), and can be isolated without
phospho-enrichment41. As a result, this peptide may be uniquely positioned as a broadly targetable antigen whose expression can be modulated by CDK4/6 inhibition. As a general effect of
palbociclib treatment, TAAs derived from proteins like MLANA (MART1), PMEL (gp100), and TYR, among others, also increased in presentation following treatment (Fig. 4i). While these antigens
and their source proteins are not universally conserved across our cell lines, the effect of increased TAA presentation following 1 μM palbociclib treatment could be applied to increase
antigen presentation prior to immunotherapies targeting these well-documented antigens. RESPONSE TO PALBOCICLIB IS REFLECTED IN THE IMMUNOPEPTIDOME To further assess whether quantitative
differences in the immunopeptidome after palbociclib treatment are reflective of the cell signaling response to a perturbation, we performed a nonparametric test to identify positively and
negatively enriched pathways. Gene names for source proteins were rank ordered according to fold change with treatment and searched against the MSigDB Hallmarks gene set database using Gene
Set Enrichment Analysis (GSEA)42,43,44. This analysis did not reveal any significantly enriched pathways for the low-dose treatment, but high-dose palbociclib showed significant enrichment
among downregulated pMHCs of E2F targets, G2M checkpoint, DNA repair, mitotic spindle, and MTORC1 signaling pathways in one or more cell lines (Fig. 5a). These findings reflect the known
biological effects of CDK4/6 inhibition. For instance, inhibiting CDK4/6 decreases expression of E2F targets, and peptides derived from E2F targets like Ki-67, a proliferation marker, were
depleted in all four cell lines (Fig. 5b). E2F also controls genes involved in DNA damage repair, and consistently, γH2AX levels, a marker of DNA double-strand breaks, increased at 72 h with
palbociclib treatment in a dose-dependent manner45 (Supplementary Fig. 4a). Although similar biological processes are enriched across the four cell lines, source proteins for significantly
depleted E2F peptides showed little overlap between the cell lines (Fig. 5b), again emphasizing the individuality of the source proteins contributing to each cell line’s detected pMHC
repertoire. Only one pathway, oxidative phosphorylation (OxPhos), was significantly upregulated in SKMEL28 cells. However, all cell lines presented peptides derived from the OxPhos pathway
that increased significantly with palbociclib treatment, although this effect was more prominent with low-dose treatment in SKMEL5, IPC298, and SKMEL2 cells, in contrast to the results of
SKMEL28 cells (Fig. 5c). OxPhos has been shown to increase with CDK4/6 inhibition due to increased ATP levels and mitochondrial mass, elevating metabolic activity. Comparably, all samples
showed elevated mitochondrial levels following treatment, suggesting that enriched pMHC presentation of OxPhos-derived peptides reflects a change in the metabolic cell state (Fig. 5d).
Because alterations to the pMHC repertoire align with previously characterized biological responses to CDK4/6 inhibition, we tested whether changes in RNA expression could predict the
quantitative immunopeptidome changes (Supplementary Data 4). No bulk correlation (_r_2 = 0.04) was observed between pMHC expression and RNA expression (Fig. 5e). This was unsurprising, as
many mechanisms beyond gene expression regulate pMHC presentation, including protein synthesis, degradation, post-transitional modifications, processing, and more. Despite this poor
correlation, significantly enriched gene sets in the immunopeptidome were also present in our RNA-sequencing (RNA-seq) analysis (Fig. 5f). While E2F pMHCs significantly depleted in SKMEL5
cells correlated with significantly decreased gene expression of the same source proteins (Supplementary Fig. 3b), only five of the 15 positively enriched OxPhos peptides displayed
significantly higher gene expression with palbociclib treatment, with three decreasing in expression, and seven remaining unchanged (Fig. 5g). Collectively, these data suggest that while
changes in gene expression and pMHC repertoires map to the same biological pathways, individual gene expression changes are not necessarily predictive of alterations in the immunopeptidome.
IFN-Γ-INDUCED PMHC ALTERATIONS ARE DISTINCT FROM PALBOCICLIB Previous work has demonstrated that CDK4/6 inhibition stimulates IFN signaling, augmenting antigen presentation levels15. We also
observed upregulation of IFN-γ response genes with low-dose palbociclib treatment, as well as increased expression of genes relating to antigen presentation (Figs. 5f, 6a). Consequently,
we tested whether direct IFN-γ stimulation would shift the repertoire similarly to CDK4/6 inhibition. Cells were stimulated with DMSO or 10 ng mL−1 IFN-γ for 72 h and the resulting pMHC
repertoires were quantified using our multiplexed hipMHC platform (Supplementary Data 5). IFN-γ increased surface pMHC levels >2× for each cell line (Fig. 6b), a trend that was reflected
in the immunopeptidome, as nearly every identified pMHC increased in presentation with stimulation (Fig. 6c, Supplementary Fig. 5a). To determine the similarity of response to palbociclib
treatment, we again performed GSEA against the hallmark gene sets. The most significantly upregulated pathway in SKMEL5 cells with IFN-γ stimulation was the “IFN-γ response,” including
peptides derived from proteins involved in antigen processing like STAT1 and HLA-A, in line with previous findings46 (Fig. 6d, e). In fact, IFN-γ response was the top enriched pathway in
every cell line, reiterating that the cellular response to stimulus is reflected in quantitative differences in pMHC presentation, and that IFN-γ-related peptides are preferentially
upregulated by IFN-γ stimulation (Supplementary Fig. 5b). Other pathways such as G2M checkpoint and mitotic spindle were positively enriched in IFN-γ stimulated cells, in contrast to the
results of palbociclib treatment. Although the cell lines showed differential pMHC pathway enrichment upon CDK4/6 inhibition with palbociclib and IFN-γ stimulation, we tested whether any
pMHCs or source proteins were commonly enriched in response to these perturbations. In SKMEL5 cells, we identified just 20 peptides and 31 source proteins significantly enriched in both
conditions (Fig. 6f, g), which primarily map to the cytoplasm and contain multiple ribosomal and translation initiation proteins frequently overrepresented in immunopeptidomic data sets
(i.e., DRiPs)47 (Fig. 6h). These data demonstrate that while CDK4/6 inhibition may induce an IFN-γ response, stimulating cells with IFN-γ does not recapitulate the distinct peptide
repertoire alterations observed with palbociclib treatment. Instead, IFN-γ stimulation alters the repertoire by augmenting the presentation of IFN-γ-related peptides. DISCUSSION The addition
of hipMHCs as internal standards improves relative quantitative accuracy for both LF and multiplexed, labeled analyses, although multiplexed labeling with TMT showed superior accuracy and
peptide binding specificity and yielded a higher number of quantifiable unique peptides using equivalent sample input. These internal hipMHC standards, which travel through the entire pMHC
workflow, also account for variation across samples and provide an estimate for dynamic range suppression, which varies across peptides. We demonstrate that hipMHC correction alters the
biological interpretation of quantitative pMHC repertoire changes, even in a relatively simple, in vitro system. Utilizing hipMHCs will be increasingly beneficial in accounting for variation
in sample losses across heterogeneous in vivo samples, and in large studies to compare and correct quantitation across many multiplexed analyses or clinical sites. While we use TMT 6-plex
in our analyses, this method is compatible with other isobaric labeling strategies, including iTRAQ (isobaric tags for relative and absolute quantitation), TMT 11-plex, and TMTpro, to
analyze up to 16 samples simultaneously. For rapid profiling of immunopeptidome changes, we elected to use minimal sample input, making this protocol easily translatable for in vivo-derived
tissue (e.g., clinical and animal) samples. While further reductions in sample input mirroring the amount obtained with a 14-gauge needle biopsy48 resulted in a notable decrease in the
number of unique peptides identified (Supplementary Fig. 1f), we believe advancements in the speed and sensitivity of mass spectrometers, as well as in sample preparation techniques to
reduce sample losses will enable pMHC profiling at even lower sample inputs in the future. Alternatively, using this same general platform of hipMHCs and isobaric multiplex labeling, the
sample amount could be increased and coupled with fractionation for deeper sequencing of the pMHC repertoire, including neoantigen identification49. In addition to improved relative
quantification, we also demonstrated the utility of hipMHCs for pMHC absolute quantification by generating an embedded multipoint standard curve. Using targeted MS to detect attomole levels
of antigen from just 1 × 107 cells and regressing this signal against the titrated hipMHC standard, we were able to extract accurate absolute quantification in terms of copies per cell for
two pMHC’s with ~20-fold difference in abundance. While absolute quantification is limited to just two peptides in this study, applying advanced targeted MS methods could enable the
quantitation of hundreds of peptides in a single analysis50. The ability to readily determine the absolute quantification of detectable antigens of interest without the need for a
pMHC-specific antibody will aid in targeted immunotherapy design. For instance, peptides of lower abundances may be better suited for engineered TCR-based therapies, as TCRs have been shown
to be incredibly sensitive with as few as one pMHC complex being capable of initiating detectable T cell activation51. Alternatively, antibody-based therapies targeting specific pMHCs, for
example, bi-specific T cell engagers or antibody–drug conjugates, may benefit from higher antigen expression levels, although results vary across antigen targets and antibody affinities52.
Moreover, absolute quantification of pMHC expression can help to untangle the biological relationships among antigen processing, epitope abundances, immunogenicity, and off-target toxicity
(e.g., tumor versus non-tumor abundance). It is worth noting that one existing restriction to using hipMHCs is the commercial availability of UV-mediated MHC monomers and ELISA control
reagents, which are limited to a handful of common human class I alleles. While matched allele hipMHCs are not required for normalization correction if MHC molecules are isolated using a
pan-specific antibody, they are necessary for accurate absolute quantification with embedded standard curves. An analogous technology, disulfide-stabilized HLA molecules, could be used in
place of UV-mediated exchange53. These HLA–B2M complexes show increased stability and higher exchange efficiency of lower-affinity peptides, potentially eliminating the need for an ELISA to
quantify exchange efficiency and simplifying MHC refolding to expand this protocol to other alleles and species. We applied our quantitative multiplexed hipMHC normalization to determine the
pMHC repertoire response to CDK4/6 inhibition with palbociclib treatment in melanoma. These results indicate that extracellular changes in pMHC abundance are reflective of the intracellular
response to CDK4/6 inhibition. Moreover, palbociclib treatment increased the presentation of TAAs and peptides derived from metabolic processes. Recently, high tumor antigen and metabolic
protein expression levels have been shown to be predictive of checkpoint inhibitor response in melanoma, suggesting that palbociclib could be used in conjunction with CB- or TIL-based
therapies to increase tumor immunogenicity54. As an alternate therapeutic strategy, peptide antigens whose surface expression was selectively increased by palbociclib could be utilized for
targeted immunotherapy, either alone or in combination. Indeed, the landscape of clinical trials exploring combination treatment regimens coupling checkpoint blockade with other therapies is
rapidly expanding55,56,57. Quantifying the molecular consequences of these combination regimes with our platform could provide insight into these trials and enable the informed design of
new therapeutic combinations, potentially with targeted immunotherapies. Taken together, our relative and absolute quantitative immunopeptidomic data demonstrate the utility of quantitative
immunopeptidomics in evaluating the pMHC repertoire response to therapy. The multiplexed nature of this platform allows for analyses of many samples in a short timescale, an important
feature in the context of clinical trials. Further analyses of pMHC repertoire changes will be useful in understanding the order and timing of therapies to achieve optimal success and may
enable predictions as to how to tune the immunopeptidome to be most applicable to immunotherapy targeting. METHODS HUMAN CELL LINES SKMEL5, SKMEL28, and MDA-MB-231 cell lines were obtained
from ATCC (ATCC HTB-70, ATCC HTB-72, and HTB-26, respectively) and maintained in DMEM (Dulbecco’s modified Eagle’s medium) (Corning). IPC298 and SKMEL2 cells were provided by Array Biopharma
and maintained in RPMI-1640 (Gibco) and minimum essential medium-α (Gibco), respectively. All media were supplemented with 10% fetal bovine serum (FBS) (Gibco) and 1%
penicillin/streptomycin (Gibco). Cells were routinely tested for mycoplasma contamination, and maintained in 37 °C, 5% CO2. PHENOTYPIC ASSAYS IC50 of palbociclib (Selleckchem, PD-0332991)
were determined for each cell line using CellTiter-Glo luminescent cell viability assay (Promega). Cells were seeded at density of 10,000 (SKMEL2, SKMEL28, IPC298) or 5,000 (SKMEL5) cells
per well in a 96-well plate and allowed to adhere overnight. Cells were then treated with palbociclib or DMSO as a vehicle control in fresh medium for 72 h and assayed. Data were acquired
using a Tecan plate reader Infinite 200 with Tecan icontrol version 1.7.1.12. IC50 values were calculated using a four-parameter logistic curve in Prism 8.4.1. Mitochondrial content was
measured using a fluorescent mitochondrial stain. Cells were seeded at a density of 20,000 cells per well in a 24-well plate and allowed to adhere overnight. Cells were then treated with 1
μM or 10 μM palbociclib or DMSO vehicle control in fresh medium for 72 h. Cells were assayed by incubating 200 nM of MitoTracker Green FM (Thermo Fisher) and a 1:1000 dilution of NuclearID
Red DNA stain (Enzo Life Biosciences) for 15 min in a serum-free medium at 37 °C. After staining and medium exchange, cells were imaged and analyzed using the Incucyte Live Cell Analysis
System (IncuCyte Zoom version 6.2.9200.0, Essen BioScience). The integrated intensity of MitoTracker dye was calculated for each image (_n_ = 3 experimental replicates, _n_ = 3 images per
sample) and divided by the number of cells (counted using nuclear counterstain) to determine the mitochondrial intensity per cell. A one-way analysis of variance followed by Dunnett’s
multiple comparisons statistical test was performed in Prism to compare the significance of treated cells versus vehicle DMSO control. Significance values represent multiplicity-adjusted _p_
values. FLOW CYTOMETRY For analysis of cells by flow cytometry, cells were lifted with 0.05% Trypsin-EDTA and 106 cells/mL were spun at 300 × _g_ for 3 min, washed with ice-cold
phosphate-buffered saline (PBS) supplemented with 1% FBS and 0.1% sodium azide (flow buffer), and incubated with fluorophore-conjugated antibody at 0.5 μg mL−1 in flow buffer for 30 min on
ice. After incubation, cells were washed again, and resuspended in flow buffer plus 5 μL of propidium iodide staining solution (10 μg mL−1, Invitrogen) per sample. Analyses were performed on
an LSRII (BD Biosciences) and data were analyzed using FlowJo (version 10.6.2). All antibodies were purchased from BioLegend: Alexa Fluor 488 HLA-A, -B, -C, clone W6/32 (cat. # 311413),
Alexa Fluor 488 anti-H2A.X Phospho (Ser139), clone 2F3 (cat. # 613406). The gating strategy used for all experiments is provided in Supplementary Fig. 6. UV-MEDIATED PEPTIDE EXCHANGE FOR
HIPMHCS UV-mediated peptide exchange was performed using recombinant, biotinylated Flex-T HLA-A*02:01 monomers (BioLegend), using a modified version of the commercial protocol. Briefly, 4 μL
of 500 μM peptide stock, 2 μL of Flex-T monomer, and 32 μL of 1× PBS were combined in a 96-well U-bottom plate. On ice, plates were illuminated with UV light (365 nm) for 30 min, followed
by a 30-min incubation at 37 °C protected from light. Concentration of stable complexes following peptide exchange was quantified using the Flex-T HLA class I ELISA assay (BioLegend) as per
the manufacturer’s instructions for HLA-A*02:01. ELISA results were acquired using a Tecan plate reader Infinite 200 with Tecan icontrol version 1.7.1.12. PMHC ISOLATION Cultured cells were
seeded in 10 cm plates, allowed to adhere overnight, and treated for 72 h with palbociclib, 10 ng mL−1 human recombinant IFN-γ (ProSpec Bio), or DMSO vehicle control. At the time of harvest,
cells were washed with 1× PBS, and lifted using 0.05% Trypsin-EDTA (Gibco). Cells were pelleted at 500 × _g_ for 5 min, washed twice more in 1× PBS, and pelleted again. Cells were
resuspended in 1 mL lysis buffer [20 nM Tris-HCl pH 8.0, 150 mM NaCl, 0.2 mM PMSO, 1% CHAPS, and 1× HALT Protease/Phosphatase Inhibitor Cocktail (Thermo Fisher)], followed by brief
sonication (3 × 10 s microtip sonicator pulses) to disrupt cell membranes. The lysate was cleared by centrifugation at 5000 × _g_ for 5 min and quantified using Bicinchoninic Acid Protein
Assay Kit (Pierce). pMHCs were isolated from 1 × 107 cells per condition with IP and size-exclusion filtration, as previously described58 Briefly, for each condition 0.5 mg of pan-specific
anti-human MHC class I (HLA-A, HLA-B, HLA-C) antibody [clone W6/32, Bio X Cell (cat. # BE0079)] was bound to 20 μL FastFlow Protein A Sepharose bead slurry (GE Healthcare) for 3 h rotating
at 4 °C. Beads were washed 2× with IP buffer (20 nM Tris-HCl pH 8.0, 150 mM NaCl) prior to lysate and hipMHC addition, and incubated rotating overnight at 4 °C to isolate pMHCs. Beads were
washed with 1× TBS and water, and pMHCs were eluted in 10% formic acid for 20 min at room temperature (RT). Peptides were isolated from antibody and MHC molecules using a passivated 10 K
molecule weight cut-off filters (PALL Life Science), lyophilized, and stored at −80 °C prior to analysis. PMHC LABELING WITH TMTS AND SP3 CLEANUP For labeled analyses, 100 μg of
pre-aliquoted TMT 6-plex (TMT) was resuspended in 30 μL anhydrous acetonitrile (MeCN), and lyophilized peptides were resuspended in 100 μL 150 mM triethylammonium bicarbonate and 50%
ethanol. Both were gently vortexed, centrifuged at 13,400 × _g_ for 1 min, and combined. TMT/peptide mixtures were incubated on a shaker for 1 h at RT, followed by 15 min of vacuum
centrifugation. After combining labeled samples, we washed tubes 2× with 25% MeCN in 0.1% acetic acid (AcOH) and added it to the labeled mixture, which was subsequently centrifuged to
dryness. Sample cleanup was performed using single-pot solid-phase-enhanced sample preparation (SP3) as previously described59. Briefly, a 1:1 mix of hydrophobic/hydrophilic Sera-mag
carboxylate-modified speed beads (GE Healthcare) was prepared at a final bead concentration of 10 μg μL−1. Labeled samples were resuspended in 30 μL of 100 mM ammonium bicarbonate (pH 7–8)
and added to 500 μg of bead mix with 1 mL MeCN. Peptides were allowed to bind for 10 min at RT, washed 2× with MeCN, and eluted with 2% DMSO for 1 min of sonication in a bath sonicator.
TMT-labeled peptides were transferred to a fresh microcentrifuge tube and centrifuged to dryness. SYNTHETIC PEPTIDE STANDARDS Heavy leucine-containing peptides were synthesized at the MIT
Biopolymers and Proteomics Lab using standard Fmoc chemistry using an Intavis model MultiPep peptide synthesizer with HATU activation and 5 μmol chemistry cycles. Starting resin used was
Fmoc-Amide Resin (Applied Biosystems). Cleavage from resin and simultaneous amino acid side chain deprotection was accomplished using: trifluoroacetic acid (81.5% v/v); phenol (5% v/v);
water (5% v/v); thioanisole (5% v/v); 1,2-ethanedithiol (2.5% v/v); 1% triisopropylsilane for 1.5 h. Standard Fmoc amino acids were procured from NovaBiochem and Fmoc-Leu (13C6, 15N) was
obtained from Cambridge Isotope Laboratories. Peptides were quality controlled by MSy and reverse phase chromatography using a Bruker MiroFlex MALDI-TOF and Agilent model 1100 HPLC system
with a Vydac C18 column (300 Å, 5 μm, 2.1 × 150 mm2) at 300 μL/min monitoring at 210 and 280 nm with a trifluoroacetic acid/H2O/MeCN mobile phase survey gradient. All peptides contain
C-terminal amidation, with the exception of the BCAP31 and DDX5 peptides used for absolute quantification. For amidated peptides, we observe C-terminal amidation and C-terminal carboxyl
groups on peptides synthesized with an amide group. Therefore, both are considered in downstream analyses. RNA-SEQUENCING RNA was isolated from 10 cm plates of SKMEL5 cells with three
biological replicates per condition. Prior to harvest, cells were washed with ice-cold 1× PBS over ice and lysed in TRIzol reagent (Thermo Fisher). Total RNA was isolated from each sample
using Direct-zol RNA MiniPrep kit (Zymo Research) according to the manufacturer’s instructions. RNAs were confirmed for quality using the Agilent Fragment Analyzer and 300 ng of material was
polyA selected using NEBNext Poly(A) mRNA Magnetic Isolation Module (E7490) modified to include two rounds of polyA binding and 10 min incubations. cDNA was generated using the NEB Ultra II
Directional Kit (E7760) following the manufacturer’s protocol using 12 cycles of PCR and a 0.9X SPRI clean. The resulting libraries were quality assessed using the Fragment Analyzer and
quantified by quantitative PCR prior to be sequenced on the Illumina HiSeq2000. The 40 nt single-end reads with an average depth of five million reads per sample were sequenced for all
conditions. RNA-seq reads were aligned to the human transcriptome prepared with the hg38 primary assembly and the Ensembl version 95 annotation using STAR version 2.5.3a60. Gene expression
was summarized with RSEM version 1.3.0 and SAMtools version 1.3 (refs61,62). Differential expression analysis was performed with DESeq2 version 1.24.0 running under R version 3.6.0 with
normal log fold change shrinkage63. Significance values (adjusted _p_ value) are determined using the Wald test, and are multiple hypothesis corrected using Benjamini–Hochberg method. The
resulting data were parsed and assembled using Tibco Spotfire Analyst version 7.11.1. MS DATA ACQUISITION For MS analysis, peptides were resuspended in 0.1% AcOH and loaded on a precolumn
packed in-house [100 μm ID × 10 cm packed with 10 μm C18 beads (YMC gel, ODS-A, 12 nm, S-10 μm, AA12S11)]. The precolumn was then washed with 0.1% AcOH and connected in series to an
analytical capillary column with an integrated electrospray tip (~1 μm orifice) with 5 μM C18 beads, prepared in-house [(50 μm ID × 12 cm with 5 μm C18 beads (YMC gel, ODS-AQ, 12 nm, S-5 μm,
AQ12S05)]. Peptides were eluted using a 130-min gradient with 10–45% buffer B (70% MeCN, 0.2 M AcOH) from 5 to 100 min and 45–55% buffer B from 100 to 120 min at a flow rate of 0.2 mL/min
for a flow split of ~10,000:1. Peptides were analyzed using a Thermo Fisher Q Exactive HF-X Hybrid Quadrupole-Orbitrap mass spectrometer, and data were acquired using Thermo Fisher
Scientific Xcalibur version 2.9.0.2923. Standard MS parameters were as follows: spray voltage, 2.5 kV; no sheath or auxiliary gas flow; heated capillary temperature, 250 °C. The HF-X was
operated in DDA mode for LF and TMT analyses. LF: Full-scan MS spectra (mass/charge ratio (_m/z_), 350–2000; resolution, 60,000) were detected in the Orbitrap analyzer after accumulation of
ions at 3e6 target value with a maximum injection time (IT) of 50 ms. For every full scan, the top 20 most intense ions were isolated (isolation width of 0.4 _m_/_z_) and fragmented
(collision energy: 28%) by higher energy collisional dissociation with a maximum injection time of 300 ms, automatic gain control target 1e5, and 60,000 resolution. Charge states <2 and
>4 were excluded, and dynamic exclusion was set to 30 s. TMT: Full-scan MS spectra (_m/z_, 400–2000; resolution, 120,000) were detected in the Orbitrap analyzer after accumulation of ions
at 3e6 target value with a maximum IT of 50 ms. For every full scan, the 20 most intense ions were isolated (isolation width of 0.4 _m_/_z_) and fragmented (collision energy: 29%) by higher
energy collisional dissociation with a maximum injection time of 350 ms, AGC target 1e5, and 30,000 resolution. Charge states <2 and >4 were excluded, and dynamic exclusion was set to
60 s. To ensure fragmentation of normalization standards, one fraction may be analyzed using targeted selected ion monitoring used in tandem with DDA with an inclusion list of hipMHC
standards. For absolute quantification, the HF-X was operated in DDA mode with inclusion list enabled. Parameters mirror those of the TMT DDA method, with several exceptions. Full-scan mass
spectra _m_/_z_ range: 300–1200, maximum MS2 injection time 200 ms, only charge states of 2 and 3 were considered. Inclusion list masses and charge states listed in Supplementary Data 6. MS
SEARCH SPACE AND FILTERING All mass spectra were analyzed with Proteome Discoverer (PD, version 2.2) and searched using Mascot (version 2.4) against the human SwissProt database. No enzyme
was used, and variable modifications included oxidized methionine for all analyses and phosphorylated serine, threonine, and tyrosine for cell treatment analyses. Treatment analyses were
also searched against a previously published catalog of over 40,000 predicted antigenic mutations in cancer cell lines64. Heavy leucine-containing peptides were searched for separately with
heavy leucine (+7), C-terminal amidation, and methionine oxidation as dynamic modifications against a custom database of the synthetic peptide standards. All analyses were filtered with the
following criteria: search engine rank = 1, isolation interference ≤ 30%, and length between 8 and 15 amino acids. LF analyses were filtered with ion score ≥ 20, and labeled samples were
filtered with ion score ≥15 and percolator _q_ value ≤ 0.05. AUC quantitation was performed using the minora feature detector in PD with match between runs enabled and filtered for ion score
≥20. For targeted, absolute quantification analyses, total ion count values for each scan and peak intensities were extracted using Skyline (version 19.1.0.193)65. MS DATA ANALYSIS WITH
HIPMHC CORRECTION For LF analyses, correction parameters were determined by calculating the ratio of AUC intensities in each sample against a reference sample and taking the mean across
hipMHCs. For TMT-labeled samples, ratios against a reference channel (usually TMT126) were calculated and the median of all ratios for correction hipMHCs was used to determine the final
correction parameters. Only PSMs of heavy leucine-coded peptides with an average reporter ion intensity within 10-fold of the interquartile range of endogenous PSM reporter ion intensities
were used for correction, as we observed drift in the correction factors when PSM TMT intensities were well beyond endogenous levels. For absolute quantification analyses, correction factors
were generated as described for TMT analyses, and used to normalize maximum peak intensity values for DDX5 and BCAP31. Notably, with mean fold changes >2× between samples (e.g., IFN-γ
stimulation), in our hands hipMHCs are no longer able to correct between conditions despite narrow isolation window (0.4 _m_/_z_). This inaccuracy may be due to co-isolation, as the
calculated correction factors reflect median fold changes of endogenous peptides. In this case, we generated correction factors for each treatment condition separately. Correction factors
were applied to AUC values in LF analyses for all peptides that were quantifiable across samples. For labeled samples, ion intensities of PSMs for each unique peptide across analyses of the
same sample were summed, after which normalization factors were applied. To evaluate differences between conditions, the log2-transformed ratio of arithmetic mean intensity for drug- and
DMSO-treated samples (_n_ = 3) was calculated. To determine if peptides were significantly increasing, an unpaired, two-sided _t_ test was performed, and peptides with _p_ ≤ 0.05 were
considered significantly increasing. To evaluate which peptides were significantly enriched above the mean, treated samples were mean centered by dividing the ion intensity of each peptide
by the mean fold change across all peptides, after which a Student’s two-tailed _t_ test was performed on adjusted values. Peptides with a mean-adjusted _p_ value ≤ 0.05 were considered
significantly enriched. Mean centering was not performed on samples where the mean log2 fold change was between −0.07 and 0.07. Data analyses were performed using Matlab version R2019b, and
Microsoft Excel version 16.34. PMHC BINDING AFFINITY Binding affinity of pMHCs was estimated using NetMHCpan-4.0 against each cell line’s allelic profile28,40 (Supplementary Fig. 3k). Only
9-mers were evaluated, and the minimum predicted affinity (nM) of each peptide was used to assign peptides to their best predicted allele. The threshold for binding was set to 500 nM.
Binding motifs for the alleles were generated using 9-mers with predicted affinity <500 nM, and visualized using WebLogo 2.8.2 (ref.66). To estimate the proportion of peptides predicted
to be binders by chance, 10 sets of 2000 random 9-mers were created by selecting with equal probability any amino acid more than 8 amino acid from a protein C terminus as a start site from
human proteins in SwissProt version 2019_2, and binding affinity prediction was performed against the alleles of MDA-MB-231 cells. Data presented in Fig. 2d are a representative example.
ENRICHMENT ANALYSES For pMHC pathway enrichment analyses, gene names from peptide source proteins were extracted and rank ordered according to the average log2 fold change over DMSO-treated
cells. In cases where more than one peptide mapped to the same source protein, the maximum/minimum was chosen, depending on the directionality of enrichment analysis. For RNA-seq data, gene
sets were rank ordered according to the mean log2 fold change value with only protein encoding genes considered. We utilized GSEA 4.0.3 pre-ranked tool against the Molecular Signatures
Database hallmarks gene sets with 1000 permutations, weighted enrichment statistic (_p_ = 1), and a minimum gene size of 8 for pMHC analyses and 15 for RNA-seq42,43,44. Results were filtered
for false discovery rate _q_ value ≤0.25, and nominal _p_ value ≤ 0.05. Significantly enriched peptides (mean-adjusted _p_ value ≤0.05) were analyzed using STRING v.11 for GO term
enrichment against biological processes and cellular components data sets67,68. Enriched categories were filtered according to false discovery rate _q_ value ≤0.05. REPORTING SUMMARY Further
information on research design is available in the Nature Research Reporting Summary linked to this article. DATA AVAILABILITY RNA-sequencing data have been deposited into the NCBI Gene
Expression Omnibus GSE144373. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier
PXD017407. Analyzed mass spectrometry immunopeptidomics data from analyses in Figs. 2–6 is available in Supplementary Data 1, 2, 3, and 4, respectively. File maps linking files to
corresponding data sets in the manuscript is available in Supplementary Data 1–3, 5. Analyzed RNA-seq data is available in Supplementary Data 3. The list of targeted masses used in Fig. 3
for absolute quantification of peptide MHCs is listed in Supplementary Data 6. The source data underlining Figs. 2d–h, 3a, c, d, 4a, d, e, h, 5a, d–g, 6b–d and Supplementary Figs. 1g, 3b, d,
e, j, and 5a, b are provided as a Source Data File. All other data are available from the corresponding author on reasonable request. REFERENCES * Caron, E. et al. The MHC I immunopeptidome
conveys to the cell surface an integrative view of cellular regulation. _Mol. Syst. Biol._ 7, 533 (2011). Article PubMed PubMed Central CAS Google Scholar * Sharma, P., Hu-Lieskovan,
S., Wargo, J. A. & Ribas, A. Primary, adaptive, and acquired resistance to cancer iImmunotherapy. _Cell_ 168, 707–723 (2017). Article CAS PubMed PubMed Central Google Scholar *
Martins, F. et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. _Nat. Rev. Clin. Oncol._ 16, 563–580 (2019). Article CAS PubMed Google
Scholar * Reits, E. A. et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. _J. Exp. Med._ 203, 1259–1271
(2006). Article CAS PubMed PubMed Central Google Scholar * Brea, E. J. et al. Kinase regulation of human MHC class I molecule expression on cancer cells. _Cancer Immunol. Res._ 4,
936–947 (2016). Article CAS PubMed PubMed Central Google Scholar * Liu, W. M., Fowler, D. W., Smith, P. & Dalgleish, A. G. Pre-treatment with chemotherapy can enhance the
antigenicity and immunogenicity of tumours by promoting adaptive immune responses. _Br. J. Cancer_ 102, 115–123 (2010). Article CAS PubMed Google Scholar * Goel, S. et al. CDK4/6
inhibition triggers anti-tumour immunity. _Nature_ 548, 471–475 (2017). Article ADS CAS PubMed PubMed Central Google Scholar * Sullivan, R. J. et al. Atezolizumab plus cobimetinib and
vemurafenib in BRAF-mutated melanoma patients. _Nat. Med._ 25, 929–935 (2019). Article CAS PubMed Google Scholar * Ascierto, P. A. et al. Dabrafenib, trametinib and pembrolizumab or
placebo in BRAF-mutant melanoma. _Nat. Med._ 25, 941–946 (2019). Article CAS PubMed Google Scholar * Hunt, D. et al. Characterization of peptides bound to the class I MHC molecule
HLA-A2.1 by mass spectrometry. _Science_ 255, 1261–1263 (1992). Article ADS CAS PubMed Google Scholar * Bassani-Sternberg, M., Pletscher-Frankild, S., Jensen, L. J. & Mann, M. Mass
spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. _Mol. Cell. Proteom._ 14, 658–673 (2015). Article
CAS Google Scholar * Khodadoust, M. S. et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. _Nature_ 543, 723–727 (2017). Article ADS CAS
PubMed PubMed Central Google Scholar * Bogunovic, B., Srinivasan, P., Ueda, Y., Tomita, Y. & Maric, M. Comparative quantitative mass spectrometry analysis of MHC class II-associated
peptides reveals a role of GILT in formation of self-peptide repertoire. _PLoS ONE_ 5, 10599 (2010). Article ADS CAS Google Scholar * Shetty, V. et al. Quantitative immunoproteomics
analysis reveals novel MHC class I presented peptides in cisplatin-resistant ovarian cancer cells. _J. Proteom._ 75, 3270–3290 (2012). Article CAS Google Scholar * Jensen, S. M., Potts,
G. K., Ready, D. B. & Patterson, M. J. Specific MHC-I peptides are induced using PROTACs. _Front. Immunol_. 9, 2697 (2018). * Loffler, M. W. et al. Mapping the HLA ligandome of
colorectal cancer reveals an imprint of malignant cell transformation. _Cancer Res._ 78, 4627–4641 (2018). Article PubMed PubMed Central Google Scholar * Murphy, J. P. et al. Multiplexed
relative quantitation with isobaric tagging mass spectrometry reveals class I major histocompatibility complex ligand dynamics in response to doxorubicin. _Anal. Chem._ 91, 5106–5115
(2019). Article CAS PubMed PubMed Central Google Scholar * Schittenhelm, R. B., Sian, T. C. C. L. K., Wilmann, P. G., Dudek, N. L. & Purcell, A. W. Revisiting the Arthritogenic
Peptide Theory: quantitative not qualitative changes in the peptide repertoire of HLA-B27 allotypes. _Arthritis Rheumatol._ 67, 702–713 (2015). Article CAS PubMed Google Scholar *
Hassan, C. et al. Accurate quantitation of MHC-bound peptides by application of isotopically labeled peptide MHC complexes. _J. Proteom._ 109, 240–244 (2014). Article CAS Google Scholar *
Wang, Q. et al. Direct detection and quantification of neoantigens. _Cancer Immunol. Res._ 7, 1748–1754 (2019). Article PubMed PubMed Central CAS Google Scholar * Hogan, K. T. et al.
Use of selected reaction monitoring mass spectrometry for the detection of specific MHC class I peptide antigens on A3 supertype family members. _Cancer Immunol. Immunother._ 54, 359–371
(2005). Article CAS PubMed Google Scholar * Tan, C. T., Croft, N. P., Dudek, N. L., Williamson, N. A. & Purcell, A. W. Direct quantitation of MHC-bound peptide epitopes by selected
reaction monitoring. _Proteomics_ 11, 2336–2340 (2011). Article CAS PubMed Google Scholar * Wu, T. et al. Quantification of epitope abundance reveals the effect of direct and
cross-presentation on influenza CTL responses. _Nat. Commun_. 10, 2846 (2019). * Bozzacco, L. et al. Mass spectrometry analysis and quantitation of peptides presented on the MHC II molecules
of mouse spleen dendritic cells. _J. Proteome Res._ 10, 5016–5030 (2011). Article CAS PubMed PubMed Central Google Scholar * Bijen, H. M. et al. Specific T cell responses against minor
histocompatibility antigens cannot generally be explained by absence of their allelic counterparts on the cell surface. _Proteomics_ 18, e1700250 (2018). Article CAS Google Scholar *
Yang, Y., Xiang, Z., Ertl, H. C. J. & Wilson, J. M. Upregulation of class I major histocompatibility complex antigens by interferon γ is necessary for T-cell-mediated elimination of
recombinant adenovirus-infected hepatocytes in vivo. _Proc. Natl Acad. Sci. USA_ 92, 7257–7261 (1995). Article ADS CAS PubMed PubMed Central Google Scholar * Rodenko, B. et al.
Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. _Nat. Protoc._ 1, 1120–1132 (2006). Article CAS PubMed Google Scholar * Jurtz, V. et al. NetMHCpan-4.0:
improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. _J. Immunol._ 199, 3360–3368 (2017). Article CAS PubMed Google Scholar *
Bassani-Sternberg, M. et al. Deciphering HLA-I motifs across HLA peptidomes improves neo-antigen predictions and identifies allostery regulating HLA specificity. _PLoS Comput. Biol_. 13,
e1005725 (2017). Article ADS PubMed PubMed Central CAS Google Scholar * Gloger, A., Ritz, D., Fugmann, T. & Neri, D. Mass spectrometric analysis of the HLA class I peptidome of
melanoma cell lines as a promising tool for the identification of putative tumor-associated HLA epitopes Europe PMC Funders Group. _Cancer Immunol. Immunother._ 65, 1377–1393 (2016). Article
CAS PubMed PubMed Central Google Scholar * Nyamao, R. M., Wu, J., Yu, L., Xiao, X. & Zhang, F. M. Roles of DDX5 in the tumorigenesis, proliferation, differentiation, metastasis and
pathway regulation of human malignancies. _Biochim. Biophys. Acta_ 1871, 85–98 (2019). CAS Google Scholar * Choi, Y. J. & Anders, L. Signaling through cyclin D-dependent kinases.
_Oncogene_ 33, 1890–1903 (2014). Article CAS PubMed Google Scholar * Hamilton, E. & Infante, J. R. Targeting CDK4/6 in patients with cancer. _Cancer Treat. Rev._ 45, 129–138 (2016).
Article CAS PubMed Google Scholar * Schettini, F. et al. CDK 4/6 inhibitors as single agent in advanced solid tumors. _Front. Oncol._ 8, 608 (2018). Article PubMed PubMed Central
Google Scholar * Deng, J. et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. _Cancer Discov._ 8, 216–233 (2018). Article CAS PubMed Google Scholar *
Donati, G., Montanaro, L. & Derenzini, M. Ribosome biogenesis and control of cell proliferation: p53 is not alone. _Cancer Res._ 72, 1602–1607 (2012). Article CAS PubMed Google
Scholar * Franco, J., Balaji, U., Freinkman, E., Witkiewicz, A. K. & Knudsen, E. S. Metabolic reprogramming of pancreatic cancer mediated by CDK4/6 inhibition elicits unique
vulnerabilities. _Cell Rep._ 14, 979–990 (2016). Article CAS PubMed PubMed Central Google Scholar * Zarling, A. L. et al. Identification of class I MHC-associated phosphopeptides as
targets for cancer immunotherapy. _Proc. Natl Acad. Sci. USA_ 103, 14889–14894 (2006). Article ADS CAS PubMed PubMed Central Google Scholar * Zarling, A. L. et al. MHC-restricted
phosphopeptides from insulin receptor substrate-2 and CDC25b offer broad-based immunotherapeutic agents for cancer. _Cancer Res._ 74, 6784–6795 (2014). Article CAS PubMed PubMed Central
Google Scholar * Scholtalbers, J. et al. TCLP: an online cancer cell line catalogue integrating HLA type, predicted neo-epitopes, virus and gene expression. _Genome Med._ 7, 118 (2015).
Article PubMed PubMed Central CAS Google Scholar * Bassani-Sternberg, M. et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by
mass spectrometry. _Nat. Commun._ 7, 13404 (2016). Article ADS CAS PubMed PubMed Central Google Scholar * Mootha, V. K. et al. PGC-1α-responsive genes involved in oxidative
phosphorylation are coordinately downregulated in human diabetes. _Nat. Genet._ 34, 267–273 (2003). Article CAS PubMed Google Scholar * Subramanian, A. et al. Gene set enrichment
analysis: a knowledge-based approach for interpreting genome-wide expression profiles. _Proc. Natl Acad. Sci. USA_ 102, 15545–15550 (2005). Article ADS CAS PubMed PubMed Central Google
Scholar * Liberzon, A. et al. The Molecular Signatures Database hallmark gene set collection. _Cell Syst._ 1, 417–425 (2015). Article CAS PubMed PubMed Central Google Scholar *
Hashizume, R. et al. Inhibition of DNA damage repair by the CDK4/6 inhibitor palbociclib delays irradiated intracranial atypical teratoid rhabdoid tumor and glioblastoma xenograft regrowth.
_Neuro Oncol._ 18, 1519–1528 (2016). CAS PubMed PubMed Central Google Scholar * Chong, C. et al. High-throughput and sensitive immunopeptidomics platform reveals profound interferon
γ-mediated remodeling of the human leukocyte antigen (HLA) ligandome. _Mol. Cell. Proteom._ 17, 533–548 (2018). Article CAS Google Scholar * Bourdetsky, D., Schmelzer, C. E. H. &
Admon, A. The nature and extent of contributions by defective ribosome products to the HLA peptidome. _Proc. Natl. Acad. Sci. USA_ 111, E1591–E1599 (2014). * Labots, M. et al.
Phosphotyrosine-based-phosphoproteomics scaled-down to biopsy level for analysis of individual tumor biology and treatment selection. _J. Proteom._ 162, 99–107 (2017). Article CAS Google
Scholar * Purcell, A. W., Ramarathinam, S. H. & Ternette, N. Mass spectrometry-based identification of MHC-bound peptides for immunopeptidomics. _Nat. Protoc._ 14, 1687–1707 (2019).
Article CAS PubMed Google Scholar * Gallien, S., Yoon Kim, S. & Domon, B. Large-scale targeted proteomics using internal standard triggered-parallel reaction monitoring (IS-PRM).
_Mol. Cell. Proteom._ 14, 1630–1644 (2015). Article CAS Google Scholar * Huang, J. et al. A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in
CD4 + T Cells. _Immunity_ 39, 846–857 (2013). Article CAS PubMed Google Scholar * Ellerman, D. Bispecific T-cell engagers: towards understanding variables influencing the in vitro
potency and tumor selectivity and their modulation to enhance their efficacy and safety. _Methods_ 154, 102–117 (2019). Article CAS PubMed Google Scholar * Moritz, A. et al.
High-throughput peptide-MHC complex generation and kinetic screenings of TCRs with peptide-receptive HLA-A*02:01 molecules. _Sci. Immunol_. 4, eaav0860 (2019). Article CAS PubMed Google
Scholar * Harel, M. et al. Proteomics of melanoma response to immunotherapy reveals mitochondrial dependence. _Cell_ 179, 236–250.e18 (2019). Article CAS PubMed PubMed Central Google
Scholar * Esteva, F. J., Hubbard-Lucey, V. M., Tang, J. & Pusztai, L. Immunotherapy and targeted therapy combinations in metastatic breast cancer. _Lancet Oncol._ 20, e175–e186 (2019).
Article CAS PubMed Google Scholar * Yu, C. et al. Combination of immunotherapy with targeted therapy: theory and practice in metastatic melanoma. _Front. Immunol._ 10, 990 (2019). *
McGranahan, T., Therkelsen, K. E., Ahmad, S. & Nagpal, S. Current state of immunotherapy for treatment of glioblastoma. _Curr. Treat. Opt. Oncol._ 20, 24 (2019). * Jaeger, A. M. et al.
Rebalancing protein homeostasis enhances tumor antigen presentation. _Clin. Cancer Res._ 25, 6392–6405 (2019). Article PubMed PubMed Central CAS Google Scholar * Browne, C. M. et al. A
chemoproteomic strategy for direct and proteome-wide covalent inhibitor target-site identification. _J. Am. Chem. Soc._ 141, 191–203 (2019). Article CAS PubMed Google Scholar * Dobin, A.
et al. STAR: Ultrafast universal RNA-seq aligner. _Bioinformatics_ 29, 15–21 (2013). Article CAS PubMed Google Scholar * Li, H. et al. The Sequence Alignment/Map format and SAMtools.
_Bioinformatics_ 25, 2078–2079 (2009). Article PubMed PubMed Central CAS Google Scholar * Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or
without a reference genome. _BMC Bioinform._ 12, 323 (2011). Article CAS Google Scholar * Anders, S. & Huber, W. Differential expression analysis for sequence count data. _Genome
Biol._ 11, R106 (2010). Article CAS PubMed PubMed Central Google Scholar * Boegel, S., Löwer, M., Bukur, T., Sahin, U. & Castle, J. C. A catalog of HLA type, HLA expression, and
neoepitope candidates in human cancer cell lines. _Oncoimmunology_ 3, e954893 (2014). * MacLean, B. et al. Skyline: an open source document editor for creating and analyzing targeted
proteomics experiments. _Bioinformatics_ 26, 966–968 (2010). Article CAS PubMed PubMed Central Google Scholar * Crooks, G. E., Hon, G., Chandonia, J. M. & Brenner, S. E. WebLogo: a
sequence logo generator. _Genome Res._ 14, 1188–1190 (2004). Article CAS PubMed PubMed Central Google Scholar * Szklarczyk, D. et al. STRING v11: protein–protein association networks
with increased coverage, supporting functional discovery in genome-wide experimental datasets. _Nucleic Acids Res._ 47, D607–D613 (2019). Article CAS PubMed Google Scholar * Ashburner,
M. et al. Gene ontology: tool for the unification of biology. _Nat. Genet._ 25, 25–29 (2000). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We
thank Array Biopharma for providing melanoma cell lines; Cristine Devlin and Michael Birnbaum for providing recombinant MHC monomers; Joe Card (Marto lab, Dana-Farber) for kindly providing
guidance on the SP3 protocol; the MIT BioMicro Center (Stuart Levine) and the Swanson Biotechnology Center for technical support, specifically the Flow Cytometry (Glenn Paradis), Biopolymers
& Proteomics (Richard Cook and Antonius Koller), and the Barbara K. Ostrom (1978) Bioinformatics (Charlie Whittaker) core facilities. This research was supported by funding from NIH
Grants U54 CA210180, P30 CA14051, Melanoma Research Alliance Team Science Award # 565436, the MIT Center for Precision Cancer Medicine, the Koch Institute Frontier Research Program through
the Kathy and Curt Marble Cancer Research Fund, and the Takeda Immune Oncology Research Fund. L.E.S. is supported by an NIH Training Grant in Environmental Toxicology (# T32-ES007020).
AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA, 02142, USA Lauren E. Stopfer, Brian A.
Joughin, Douglas A. Lauffenburger & Forest M. White * Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA, 02139, USA Lauren E. Stopfer, Joshua M.
Mesfin, Brian A. Joughin, Douglas A. Lauffenburger & Forest M. White Authors * Lauren E. Stopfer View author publications You can also search for this author inPubMed Google Scholar *
Joshua M. Mesfin View author publications You can also search for this author inPubMed Google Scholar * Brian A. Joughin View author publications You can also search for this author inPubMed
Google Scholar * Douglas A. Lauffenburger View author publications You can also search for this author inPubMed Google Scholar * Forest M. White View author publications You can also search
for this author inPubMed Google Scholar CONTRIBUTIONS L.E.S. and F.M.W. conceived the project. F.M.W. and D.A.L. advised the project. L.E.S. and J.M.M. performed experiments. L.E.S. and
B.A.J. analyzed data. L.E.S., B.A.J., and F.M.W. wrote the manuscript, and J.M.M. and D.A.L. provided manuscript feedback. CORRESPONDING AUTHOR Correspondence to Forest M. White. ETHICS
DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PEER REVIEW INFORMATION _Nature Communications_ thanks Étienne Caron and the other,
anonymous, reviewer(s) for their contribution to the peer review of this work. PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and
institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 SUPPLEMENTARY DATA 3
SUPPLEMENTARY DATA 4 SUPPLEMENTARY DATA 5 SUPPLEMENTARY DATA 6 REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original
author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the
article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use
is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Stopfer, L.E., Mesfin, J.M., Joughin, B.A. _et al._ Multiplexed relative and
absolute quantitative immunopeptidomics reveals MHC I repertoire alterations induced by CDK4/6 inhibition. _Nat Commun_ 11, 2760 (2020). https://doi.org/10.1038/s41467-020-16588-9 Download
citation * Received: 18 February 2020 * Accepted: 13 May 2020 * Published: 02 June 2020 * DOI: https://doi.org/10.1038/s41467-020-16588-9 SHARE THIS ARTICLE Anyone you share the following
link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature
SharedIt content-sharing initiative