Synergism between il7r and cxcr4 drives bcr-abl induced transformation in philadelphia chromosome-positive acute lymphoblastic leukemia
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Ph+ acute lymphoblastic leukemia (ALL) is characterized by the expression of an oncogenic fusion kinase termed BCR-ABL1. Here, we show that interleukin 7 receptor (IL7R) interacts
with the chemokine receptor CXCR4 to recruit BCR-ABL1 and JAK kinases in close proximity. Treatment with BCR-ABL1 kinase inhibitors results in elevated expression of IL7R which enables the
survival of transformed cells when IL7 was added together with the kinase inhibitors. Importantly, treatment with anti-IL7R antibodies prevents leukemia development in xenotransplantation
models using patient-derived Ph+ ALL cells. Our results suggest that the association between IL7R and CXCR4 serves as molecular platform for BCR-ABL1-induced transformation and development
of Ph+ ALL. Targeting this platform with anti-IL7R antibody eliminates Ph+ ALL cells including those with resistance to commonly used ABL1 kinase inhibitors. Thus, anti-IL7R antibodies may
provide alternative treatment options for ALL in general and may suppress incurable drug-resistant leukemia forms. SIMILAR CONTENT BEING VIEWED BY OTHERS INTERLEUKIN-7 RECEPTOR Α MUTATIONAL
ACTIVATION CAN INITIATE PRECURSOR B-CELL ACUTE LYMPHOBLASTIC LEUKEMIA Article Open access 14 December 2021 ENDOGENOUS IL-1 RECEPTOR ANTAGONIST RESTRICTS HEALTHY AND MALIGNANT
MYELOPROLIFERATION Article Open access 03 January 2023 AN INSTRUCTIVE ROLE FOR INTERLEUKIN-7 RECEPTOR Α IN THE DEVELOPMENT OF HUMAN B-CELL PRECURSOR LEUKEMIA Article Open access 03 February
2022 INTRODUCTION The Philadelphia chromosome (Ph) is the most frequent abnormality among adults with acute lymphoblastic leukemia (ALL) (25–30%) and results in _BCR-ABL1_ fusion gene1.
Furthermore, 3–5% of children harbor this translocation which is associated with a poor prognosis2,3. As this oncogene confers constitutive kinase activity, addition of tyrosine kinase
inhibitors (TKIs) such as imatinib mesylate to intensive chemotherapy has improved the outcome of BCR-ABL1-positive leukemia to a 5-year disease-free survival rate in children (70 ± 12%, _n_
= 28)3. Nevertheless, Ph+ ALL patients still suffer from poor prognosis in both children and adults as relapse frequently occurs after stem cell transplantation. A deep understanding of the
molecular mechanisms which are associated with BCR-ABL1 transformation is of high importance in order to provide better treatment for these patients and to overcome TKI-resistance.
Recently, our group has shown that interleukin 7 receptor (IL7R) is widely expressed in B cell precursor-ALL (BCP-ALL), and that high expression levels of IL7R are correlated with central
nervous system involvement (CNS) and may predict CNS-relapse4. The cytokine IL7 binds to IL7Rα chain that hetero-dimerizes with the common gamma chain (γc) to form the IL7 receptor and
induces the kinase activity of JAK1/JAK35. Alternatively, the IL7Rα chain hetero-dimerizes with the cytokine receptor-like factor 2 (CRLF2) to form the thymic stromal lymphopoietin receptor
and mediate activation of JAK1/JAK26. The constitutive expression of IL7R7,8 in ALL together with the high frequency of mutations affecting IL7R signaling point to a key role of IL7R in
disease pathogenesis9,10,11. Thus, investigating the regulation of IL7R function is important for understanding its role in the pathogenesis of ALL. Moreover, characterizing the molecular
interaction of IL7R might provide crucial insights into the mechanisms of malignant transformation. Available data suggest that IL7R expression is controlled by the Forkhead box
transcription factor 1 (FOXO1) in lymphocytes12. Importantly, FOXO1 is essential during early B cell development and its activity is negatively regulated by phosphatidylinositol-3-kinase
(PI3K) signaling13. Therefore, FOXO1 function depends on the lipid phosphatase PTEN (phosphatase and tensin homolog) which counteracts PI3K function14. The C-X-C chemokine receptor 4 (CXCR4)
is a G-protein-coupled receptor which is widely expressed on hematopoietic stem cells and hematopoietic cancers. Together with its ligand CXCL12 (also known as stromal-derived factor 1),
CXCR4 plays an important role in tumorigenesis by regulating survival, migration, homing, and interaction of leukemia cells with their microenvironment15. High CXCR4 protein expression is
correlated with an increased risk of relapse and poor outcome in pediatric ALL patients16. Interestingly, CXCL12 was initially identified as a soluble factor that collaborates with IL7 to
activate the proliferation of progenitor B cells17,18. In this study we have investigated the molecular mechanisms, which are regulated by the oncogenic kinase BCR-ABL1 and are required for
malignant transformation or for rescue from kinase inhibitor treatment. We show that IL7R and CXCR4 interact on the cell surface and that both are crucial for malignant transformation of
early B cells by BCR-ABL1. Importantly, we show that anti-IL7R antibody can efficiently eliminate inhibitor-resistant Ph+ patient ALL in preclinical xenograft model. RESULTS BCR-ABL1 ALTERS
IL7R AND CXCR4 REGULATED GENES EXPRESSION To better understand the molecular mechanisms regulating BCR-ABL1-induced transformation and the development of Ph+ ALL, we performed RNA-sequencing
(RNA-Seq) and compared transcriptome profile of transformed cells with wildtype (WT) pre-B cells. To this end, six individually generated control WT pre-B cell lines and six
BCR-ABL1-transformed pre-B cell counterparts were analyzed. Global transcription profile based principal component analysis (PCA) showed clear segregation of WT and BCR-ABL1-transformed
cells (cumulative explained variance = 86.1%; Supplementary Fig. 1a). Gene Ontology (GO) analysis for biological processes was performed and genes which were differentially regulated between
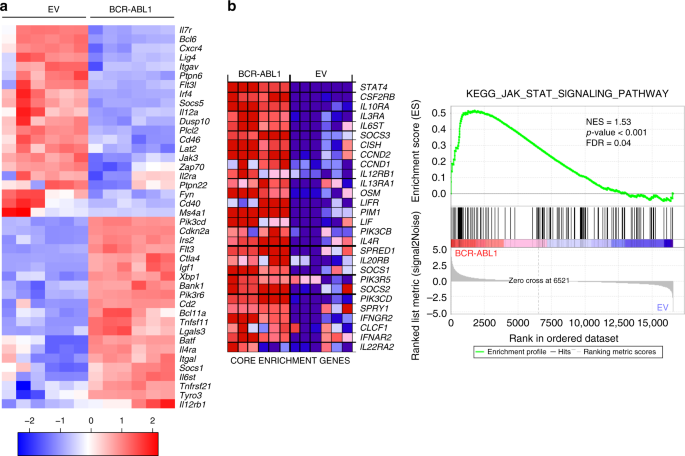
the two groups were further investigated, particularly genes related to lymphocyte activation, proliferation, and migration (Fig. 1a and Supplementary Data 1). The analysis showed a
differential regulation in multiple signaling pathways related to IL7R signaling (Fig. 1a and Supplementary Fig. 1b). To assess the importance of IL7R signaling related pathways and
processes in BCR-ABL1, we performed Gene Set Enrichment Analysis (GSEA) on IL7R related KEGG and REACTOME MSigDB gene sets (Broad Institute, Inc., Massachusetts Institute of Technology
(MIT), and Regents of the University of California). Of the eight gene sets analyzed, five showed statistically significant upregulation in BCR-ABL1 as compared with control samples (false
discovery rate (FDR) < 0.25; Fig. 1b, Supplementary Fig. 1b, and Supplementary Table 1). Interestingly, genes involved in JAK/STAT signaling, signaling by interleukins, and cytokine
signaling were among the most significantly altered gene sets (Fig. 1b and Supplementary Fig. 1b). CXCR4 pathway, though not statistically significant, showed positive correlation to the
BCR-ABL1-transformed samples (Supplementary Fig. 2). In addition, the expression of several cytokine signaling regulators such as the transcription repressor _Bcl6_ as well as several
phosphatases including _Ptpn6_, _Ptpn22_, and _Dusp10_ were deregulated by BCR-ABL1 (Fig. 1a). In this work, we focused on the role of IL7R and CXCR4. IL7 RESCUES BCR-ABL1+ CELLS FROM
INHIBITOR TREATMENT Our data suggest that the signaling pathways of IL7R and CXCR4 are tightly regulated by the activity of the oncogenic kinase BCR-ABL1 and therefore we hypothesized that
they might be directly involved in malignant transformation. To test whether the expression of IL7R and CXCR4 is also correlated in primary ALL, we analyzed a cohort of 68 Ph+ BCP-ALL
patients (patients’ characteristics are given in Supplementary Table 2) and found significant correlation of _IL7R_ and _CXCR4_ gene expression (Spearman _r_ = 0.6264; _p_ < 0.0001; Fig.
2a), suggesting that our sequence analysis of BCR-ABL1-transformed pre-B cells is in accordance with in vivo condition. In addition, searching in a mixed leukemia gene expression study19
using the R2 database (http://r2.amc.nl) showed that _IL7R_ and _CXCR4_ are expressed at reduced levels in BCR-ABL+ ALL (t9; 22) in comparison with other BCP-ALL entities (Supplementary Fig.
3a and Supplementary Table 3). Similar results were also observed in RNA-seq dataset of 1223 BCP-ALL patients20 (Supplementary Fig. 3b and Supplementary Table 3). Interestingly, the
inhibition of BCR-ABL1 kinase by imatinib treatment resulted in an upregulated expression of both the chemokine receptor CXCR4 and the IL7R together with downstream signaling elements such
as _Jak1_ and _Stat5a_ (Fig. 2b, c, and Supplementary Fig. 4a). To test whether the upregulation of IL7R or CXCR4 expression upon BCR-ABL1 kinase inhibition affects the survival of
BCR-ABL1-transformed cells, we investigated imatinib-induced cell death in the presence of the respective cytokine/chemokine. We found that treatment with IL7 counteracted imatinib-induced
cell death and restored the cell cycle progression (Fig. 2d and Supplementary Fig. 4b). However, treatment with CXCL12, the ligand for CXCR4, or its antagonist AMD3100 did not affect
imatinib treatment (Fig. 2e and Supplementary Fig. 4b, c). Likewise, treatment with TSLP, the ligand for CRLF2, was also unable to rescue BCR-ABL1-transformed cells from inhibitor-induced
cell death (Supplementary Fig. 4c). Interestingly, human Ph+ ALL SUP-15 cells also upregulated IL7R and CXCR4 in response to imatinib treatment (Supplementary Fig. 4d). Together, these data
suggest that Ph+ ALL cells upregulate growth factor receptors including IL7R which might enable the survival of Ph+ cells in microenvironments containing IL7 despite ABL1 kinase inhibitor
treatment. BCR-ABL1 TRANSFORMATION REQUIRES IL7R EXPRESSION The upregulation of IL7R under imatinib treatment raised the question whether IL7R expression is required for BCR-ABL1 induced
pre-B cell transformation and ALL development. Therefore, we generated BCR-ABL1-transformed BM-derived pre-B cells from mice homozygous for _loxP_-flanked _Il7rα_ alleles (_Il7rα__fl/fl_)21.
Usually, pre-B cells proliferate in the presence of growth factors such as IL7. However, the expression of BCR-ABL1 results in growth factor-independent proliferation in the absence of IL7
(Fig. 3a, b). For inducible deletion of the _Il7rα gene_, we introduced a tamoxifen (Tam)-inducible Cre (Cre-ERT2) into the BCR-ABL1-transformed _Il7rα__fl/fl_ cells. Inducible deletion of
the _Il7rα_ gene led to cell death of the BCR-ABL1-transformed pre-B cells (Fig. 3c–e). To determine the role of IL7R expression in vivo, we injected BCR-ABL1-transformed _Il7rα__fl/fl_
pre-B cells into NOD-SCID immunodeficient recipient mice. _Il7r_ deletion by Tam treatment in vivo reduced leukemic cell burden and significantly prolonged the survival of xenograft mice
injected with BCR-ABL1-transformed cells (Fig. 3f, g). In support of these results, BM-derived cells from _Il7r__α_-deficient mice22 did not give rise to BCR-ABL1-transformed pre-B cells,
while BCR-ABL1-transformed myeloid cells (CD11b+) can readily be generated from the same cells (Supplementary Fig. 5). Together, our data suggest that IL7R expression is specifically
required for the initiation and the maintenance of pre-B cell transformation and ALL development. IL7R SYNERGIZES WITH CXCR4 FOR BCR-ABL1+ TRANSFORMATION Since CXCR4 regulated genes were
also altered, we tested whether CXCR4 is also required for BCR-ABL1-induced transformation. Therefore, we generated BCR-ABL1-transformed pre-B cells from mice homozygous for _Cxcr4
loxP_-flanked alleles23 (_Cxcr4__fl/fl_). Deleting _Cxcr4_ in these cells using Cre-ERT2 resulted in rapid cell death and inability of BCR-ABL1 cells to form colonies in vitro (Supplementary
Fig. 6). Since BCR-ABL1 was reported to be involved in crosstalk with CXCR424, we investigated whether the requirement for IL7R and CXCR4 in BCR-ABL1-induced transformation is mediated by
spatial receptor colocalization. We first examined the effect of BCR-ABL1 recruitment on CXCR4-mediated Ca2+ mobilization25. Therefore, the CXCL12-induced Ca2+ flux in WT as compared with
BCR-ABL1-transformed cells was tested. While WT cells showed a negligible CXCL12-induced Ca2+ flux, BCR-ABL1-transformed cells showed a robust Ca2+ response (Fig. 4a). Inhibiting the
BCR-ABL1 kinase activity by either imatinib or dasatinib blocked the CXCL12-induced Ca2+ response with dasatinib showing an effective inhibition at much lower concentrations than imatinib,
which is most likely caused by the additional effect of dasatinib on Src kinases26 (Fig. 4a). Importantly, inducible deletion of _Cxcr4_ in BCR-ABL1-transformed cells or treating them with
AMD3100, an antagonist of CXCL12, prevented the Ca2+ response (Supplementary Fig. 7a). These data are in full agreement with the view that BCR-ABL1 is recruited to CXCR4 and can be activated
by the respective ligand CXCL12. Interestingly, the _Cxcr4_-deficient pre-B cells showed an increased differentiation capacity as measured by the elevated ratio of cells expressing the
immunoglobulin kappa light chain (Supplementary Fig. 7b). These data suggest that CXCR4 cooperates with IL7R in preventing pre-B cell differentiation27. Similarly, IL7R seems to act together
with CXCR4 in directing cell migration, as both _Cxcr4_-defcient and _Il7r_-defcient BCR-ABL1-transformed cells show an impaired migration towards a CXCL12 gradient (Supplementary Fig. 7c).
To study further how IL7R and CXCR4 act synergistically to regulate pre-B cell differentiation and proliferation, we investigated the interaction between IL7R and CXCR4 by proximity
ligation assay (PLA). Adjacent binding of the ligands IL7 (7 kD) and CXCL12 (15 kD) suggests that the corresponding receptors are localized on the cell surface at a proximity below 10 nm in
precursor B cells (Fig. 4b and Supplementary Fig. 8a). Interestingly, BCR-ABL1-transformed pre-B cells show an increased number of IL7R/CXCR4 foci as compared with untransformed WT pre-B
cells (Fig. 4c and Supplementary Fig. 8b). This association is also detected in human BCR-ABL+ pre-B ALL cells (Supplementary Fig. 8c). The interaction between CXCR4/IL7R and BCR-ABL was
further confirmed by immunoprecipitation (Supplementary Fig. 8d). Together, these findings suggest that the interaction between IL7R and CXCR4 is increased in BCR-ABL1-transformed pre-B
cells. Hence, we postulated that this interaction recruits IL7R-associated signaling proteins into close proximity to CXCR4 thereby enabling activation by BCR-ABL1 which then leads to pre-B
cell transformation. To directly test this hypothesis, we investigated the association of JAK3 with CXCR4 in BCR-ABL1-transformed cells as compared with WT control. Usually, JAK3 is
associated with the γc subunit of IL7R but not with CXCR4. In fact, CXCL12/CXCR4 signaling was shown to be independent of JAK328. Interestingly, a significant IL7R-dependent increase in JAK3
association with CXCR4 was observed in BCR-ABL1-transformed pre-B cells (Fig. 4d). Similarly, an IL7R-dependent association of CXCR4 with pJAK3 was observed in BCR-ABL1-transformed cells
(Supplementary Fig. 8e). In contrast, the association between IL7R and JAK3 showed no significant change suggesting that BCR-ABL1-induced transformation has no effect on IL7R interaction
with its downstream signaling elements (Supplementary Fig. 8f). As expected, JAK kinases show increased phosphorylation in BCR-ABL1-transformed cells and imatinib treatment reduces this
phosphorylation (Fig. 4e). In full agreement with the hypothesis that the interaction between CXCR4 and IL7R enables CXCR4 to utilize the downstream signaling machinery of IL7R, inducible
inactivation of either IL7R or CXCR4 expression results in decreased activity of JAK1, JAK2, and JAK3 as shown by their reduced phosphorylation (Fig. 4e). Together, these data suggest that
BCR-ABL1 interaction with CXCR4 recruits this oncogene into the proximity of IL7R-associated JAK kinases thereby enabling their BCR-ABL1-mediated activation and pre-B cell transformation.
BCR-ABL1 CONTROLS IL7R EXPRESSION BY REGULATING FOXO1 Activated JAK kinases phosphorylate the cytoplasmic domain of cytokine receptors at specific tyrosine residues leading to the
recruitment and subsequent activation of signal transducer and activator of transcription (STAT) proteins. Phosphorylated STATs undergo dimerization and translocate to the nucleus where they
activate target genes involved in proliferation and survival of lymphocytes29. As expected, increased STAT5 phosphorylation was detected in BCR-ABL1-transformed cells and ABL1 kinase
activity was required for this increase (Fig. 5a). Since STAT5 is activated by IL7R signaling30, and also by BCR-ABL1, we postulated that activated STAT5 might control IL7R expression in a
negative feedback loop that prevents deregulated IL7R expression. The transcription factor FOXO1 was shown to regulate the expression of IL7R12 as well as CXCR431,32. The fact that STAT5
activates PI3K signaling33 which in turn suppresses FOXO1 transcriptional activity by phosphorylation of specific S/T sites, suggests that STAT5 activation can lead to increased FOXO1
phosphorylation and subsequent downregulation of IL7Rα expression. Indeed, imatinib treatment of BCR-ABL1-transformed pre-B cells resulted in decreased FOXO1 phosphorylation (Fig. 5a).
Moreover, introducing a constitutively active STAT5 version (STAT5-CA)34 into pre-B cells resulted in FOXO1 inactivation, as measured by increased S256 phosphorylation, and decreased IL7Rα
expression (Fig. 5b, c). In full agreement of transcriptional repression, reverse transcriptase PCR experiments revealed that _Il7r__α_ transcripts were almost missing in STAT5-CA expressing
cells (Fig. 5d). Interestingly, the reduced IL7R expression was associated with loss of the cells expressing STAT5-CA (Fig. 5e). These findings suggest that STAT5 regulates IL7R expression
in a negative feed-backloop and that fine-tuned STAT5 activity in transformed cells is important to induce cell proliferation and, at the same time, avoid destruction of IL7R expression by
excessive STAT5 activity. Altogether, we propose that BCR-ABL1 controls IL7R expression by activating a common STAT5-regulated negative feedback mechanism and that the observed
downregulation of IL7R expression by BCR-ABL1 guarantees a fine-tuned STAT5 activity. FOXO1 IS REQUIRED FOR LEUKEMOGENESIS To further confirm the requirement of FOXO1 transcription factor
for BCR-ABL1-mediated leukemogenesis, we generated BCR-ABL1-transformed pre-B cells from mice homozygous for _loxP_-flanked alleles of _FoxO1_ (_FoxO1__fl/fl_). To induce _FoxO1_ deletion,
we introduced into the BCR-ABL1-transformed cells our Tam-inducible Cre-ERT2 by retroviral transduction. Inducible deletion of _FoxO1_ led to cell loss of the BCR-ABL1-transformed cells
(Fig. 6a–c). Moreover, deletion of _FoxO1_ resulted in concomitant downregulation of FOXO1 and IL7R expression (Fig. 6d). To provide further evidence for the important role of FOXO1 in
leukemogenesis in vivo, we injected BCR-ABL1-transformed _FoxO1__fl/fl_ cells into sublethally irradiated NOD/SCID recipient mice and monitored development of leukemia after deletion of
_FoxO1_ as compared with controls in vivo. We found that BCR-ABL1-transformed _FoxO1__fl/fl_ caused fatal leukemia within 2 weeks, while deleting _FoxO1_ by Tam-induced Cre-ERT2 activation
reduced the leukemic cell burden and prolonged the survival time of respective mice (Fig. 6e, f). Together, these data suggest that FoxO1 plays an essential role in BCR-ABL1-induced
transformation most likely through the activation of IL7R expression (Supplementary Fig. 9). TARGETING IL7R PREVENTS BCR-ABL1+ LEUKEMIA DEVELOPMENT Since the above results show that IL7R is
crucial for the transforming signals initiated by BCR-ABL1 in Ph+ ALL, we investigated whether inhibition of IL7R signaling using ruxolitinib, a JAK1/JAK2 kinase inhibitor, can interfere
with the survival of BCR-ABL1-transformed cells or enhance the effect of kinase inhibitors on these cells. We found that treatment with ruxolitinib along with imatinib prevented the
IL7-driven rescue of BCR-ABL1-transformed pre-B cells in vitro (Supplementary Fig. 10a–c). To further study the consequences of ruxolitinib on leukemia cells in vivo, we injected human
BCR-ABL+ ALL cells into NSG mice and monitored the recipient mice under imatinib, ruxolitinib, or combined imatinib/ruxolitinib treatment. We found that combination treatment was unable to
prolong the survival of recipient mice or to reduce the percentage of leukemic cells in the BM and in the spleen (Supplementary Fig. 10d–f) suggesting that, in contrast to the in vitro
results, ruxolitinib cannot support imatinib in a xenograft model and therefore may not be suitable for ALL treatment in vivo. Therefore, we tested whether direct targeting of the IL7R using
specific monoclonal antibodies may interfere with its function in leukemia. To this end, we injected imatinib-resistant BCR-ABL1+ ALL patient cells35 into NSG mice and treated them with
monoclonal antibody specific for human IL7Rα4 (Fig. 7b–d). As expected, imatinib was unable to prevent leukemia development and, therefore, the leukemia burden was increased and the survival
of imatinib-treated mice was reduced similar to that of control mice (Fig. 7a, b). In contrast, anti-IL7R antibody significantly delayed leukemia onset in vivo and led to a significantly
expanded survival time of the respective animals (Fig. 7a, b and Supplementary Fig. 11a, b). Interestingly, the xenograft patient material showed an upregulation in _BCR-ABL1_ expression
compared with human BCR-ABL1+ cell lines TOM-1 and SUP-B15 (Fig. 7c) which may explain the imatinib-resistant phenotype35. Indeed, we found that regulated long-term exposure to imatinib can
lead to upregulation of _BCR-ABL1_ expression and to imatinib-resistance in vitro (Supplementary Fig. 12). The xenograft patient material which was used lacked the BCR-ABL1 gate keeper
mutation, known as T315I kinase domain mutation, which leads to resistance against ABL1 inhibitors36,37. Therefore, we repeated the experiment using T315I-positive, imatinib resistant,
xenograft patient material38. Treatment of recipient mice with anti-IL7R antibody significantly delayed leukemia onset and led to significant prolongation of the survival time of the
respective animals (Fig. 7d–f and Supplementary Fig. 11c). As NSG mice lack NK cells, we excluded that it led to cell death via antibody-dependent cell-mediated cytotoxicity as previously
described by other IL7R antibodies39. Similarly, the antibody which we used does not block IL7 binding (Supplementary Fig. 13a), alternatively, it disrupts the scaffold between IL7R and
CXCR4 (Supplementary Fig. 13b). In addition, antibody treatment seems to enhance apoptosis as shown by increased cleavage of caspase-840 (Supplementary Fig. 13c). Together, these experiments
show that IL7R plays a pivotal role in the survival of ALL and that targeting IL7R via specific antibodies exerts a profound effect on elimination of kinase inhibitor-resistant ALL in vivo.
DISCUSSION Previous studies demonstrated remarkable outcome improvements in Ph+ ALL patients upon imatinib integration into chemotherapy41. However, acquired drug resistance is still a
crucial issue that leads to relapse of the disease and unfavorable outcome3,42. A thorough understanding of the molecular mechanisms involved in BCR-ABL1-mediated transformation is required
in order to provide therapeutic alternatives for Ph+ ALL patients, particularly those who developed TKI-resistance. In this study, we employed several genetically modified systems as well as
preclinical xenograft models to better understand BCR-ABL1-induced transformation. Interestingly, our data show that BCR-ABL1 oncogene regulates the expression and function of the signaling
pathways of IL7R and CXCR4 in a concerted manner. This combined regulation is important because both receptors are required for the growth and survival of BCR-ABL1-transformed pre-B cells.
Importantly, IL7R and CXCR4 act in close proximity thereby allowing their downstream signaling pathways to synergize and enable BCR-ABL1-induced pre-B cell transformation. In this synergism,
CXCR4 attracts the oncogenic kinase BCR-ABL1 while IL7R conveys the JAK/STAT signaling machinery. Importantly, this complex seems to act in a ligand-independent manner to activate multiple
downstream signaling pathways and is required for the survival of mouse and human leukemia cells in both in vitro as well as in vivo preclinical xenograft model. Our results indicate that
BCR-ABL1 utilizes the IL7R signaling machinery for pre-B cell transformation and growth factor-independent proliferation and that the feedback regulation of this machinery is a crucial part
of the transformation process. For instance, deregulated BCR-ABL1 kinase activity may result in uncontrolled STAT5 phosphorylation and negative feedback regulation of IL7R expression leading
to cell death. Previous report suggested that BCR-ABL oncogene mimics pre-BCR signaling by activating STAT5 on one hand and repressing BCL6 expression on the other hand7. STAT5 was also
shown to directly downregulate BCL6 expression in response to IL7 stimulation43. This is in agreement with our data showing that BCR-ABL1 transformation downregulates the transcription
repressor BCL6. Thus, BCR-ABL1-mediated pre-B cell transformation requires an equilibrium between kinase activity and negative feedback regulation of IL7R signaling. In full agreement,
BCR-ABL1-transformed pre-B cells require multiple phosphatases that are most likely involved in stabilizing this equilibrium38 which can be targeted for efficient treatment of Ph+ ALL. It is
feasible that additional players participate in regulating IL7R expression in ALL. For example, it was previously shown that _IKAROS_ negatively regulates _IL7R_ promoter and that _IKAROS_
deficiency in ALL patients is correlated with increased IL7R expression44. Similarly, the common _IKZF1_ deletion leading to dominant negative IK6 isoform45 resulted in increased IL7R
expression in BCR-ABL+ cells46. Thus, co-occurring genomic alterations such as _IKZF1_ deletion remain to be addressed in future studies. Previous reports showed that combined targeting of
BCR-ABL1 and JAK2 using dasatinib and ruxolitinib, respectively, reduced leukemia engraftment and prolonged survival47. However, these mice eventually relapsed and died from leukemia which
suggest that ruxolitinib treatment is inefficient in vivo47. This is in agreement with our results showing that inhibition of the kinases JAK1/JAK2 by ruxolitinib, applied either alone or in
combination with imatinib, was not able to provide any therapeutic advantage for xenograft animal models injected with Ph+ ALL patient material. It is conceivable that reduced drug
availability or insufficient inhibition of IL7R signaling, as ruxolitinib mainly inhibits JAK1 and JAK2 while IL7R can also activate JAK3, are responsible for the inability of ruxolitinib to
block the development of Ph+ ALL in vivo. Intriguingly, our experiments point to an unpredicted escape mechanism of transformed cells during TKI treatment. Since leukemic cells maintain the
expression of growth factor receptors such as IL7R, which is used as scaffold for organizing the oncogenic signaling machinery, the presence of IL7 in certain niches might provide the
transformed cells with escape mechanisms upon treatment with inhibitors blocking BCR-ABL148. This scenario is also possible for other growth factor receptors and their respective cytokines.
For example, it has been shown that IL3 can rescue BCR-ABL+ CML cells from cell death induced by BCR-ABL inhibitors47,49. Although our data showed that several receptors were upregulated in
response to imatinib (such as IL7R, CXCR4, and CRLF2), IL7 showed a unique potential to rescue the cells under kinase inhibitor treatment. Thus, it is conceivable that, during treatment of
Ph+ALL patients with inhibitors blocking BCR-ABL1 kinase activity, IL7R-driven survival pathways in ALL cells are activated in microenvironments containing IL7 thereby enabling the survival
of ALL cells. Ph+ ALL cells that survive treatment with BCR-ABL inhibitors in microenvironments containing IL7 may act as leukemia initiating cells and disseminate to other locations when
inhibitor concentrations decline or when inhibitor resistance is induced by somatic mutations. This scenario is further supported by the elevated amounts of IL7 detected in ALL patients9,10.
Thus, understanding the molecular mechanisms of BCR-ABL1-induced transformation is important for identifying TKI escape mechanisms and for developing strategies that prevent such escape.
Our findings may also have important therapeutic implications in other leukemia subtypes with similar gene expression such as Ph-like ALL. For example, at least 90% of patients with Ph-like
ALL showed kinase-activating alterations (e.g., in _ABL1_/_ABL2_ and _JAK2_), sequence mutations in _IL7R_ as well as an activation of phosphorylated STAT550. This suggests that IL7R might
also be a potential therapeutic target for several BCP-ALL patients who are not Ph+ as well4. Nevertheless, additional work would be required to investigate whether our model also function
in Ph-like ALL. Since IL7R expression and function is critical for proper lymphopoiesis, targeting this pathway may have effects on other normal cells. For instance, previous studies showed
that mice deficient in _Il7r_ showed depletion in both B and T lymphocytes51. In humans, mutations in the _IL7Rα_ result in severe combined immunodeficiency (SCID) which is associated with
the absence of T cells and normal numbers, nevertheless inactive, B cells52. Accordingly, targeting IL7Rα using specific antibodies may also affect T cells53 and lead to immunodeficiency in
patients. However, a recent study showed that treating healthy subjects with anti-human IL7R antibody was well tolerated and did not result in obvious alterations in immune cell populations
and inflammatory cytokine profiles54. Thus, treatment with anti-IL7R antibodies might provide a key therapeutic approach especially for TKI-resistant ALL once the different antibodies are
characterized regarding their side-effects and compared with standard chemotherapy in appropriate clinical trials. METHODS PATIENT SAMPLES, HUMAN CELL LINES Sixty-eight BCR-ABL+ ALL patients
were treated according to European intergroup study of post-induction treatment of Philadelphia chromosome-positive ALL (EsPhALL) 2004 and 2010 protocols (NCT00287105) and
ALL-Berlin-Frankfurt-Münster (BFM) 2000 (NCT00430118) study. Informed consent was obtained according to institutional regulations, in accordance with the Declaration of Helsinki. 697,
SUP-B15, and TOM-1 cell lines were obtained from DSMZ. Ph+ ALL cells containing T315I mutation was kindly provided by Markus Müschen38. MICE All mouse housing, breeding, and surgical
procedures were approved by the governmental institutions of Baden-Württemberg (Regierungspräsidium Tübingen). BM cells from WT (_n_ = 7, female), _Il7r__α_Δ (_n_ = 7, female),
_Il7r__α__fl/fl_ (_n_ = 7, female), _Cxcr4__fl/fl_ (_n_ = 3, female) and _FoxO1__fl/fl_ (_n_ = 3, female) mice were collected and retrovirally transformed with either an empty pMIG vector or
with a pMIG vector expressing BCR-ABL1. Unless mentioned otherwise, cells were cultured for 3–7 days in Iscove’s medium (Biochrom AG) containing 10% heat-inactivated FCS (Sigma-Aldrich), 2
mM l-glutamine, 100 U/ml penicillin (Gibco), 100 U/ml streptomycin (Gibco), and 50 µM 2-mercaptoethanol. The medium was supplemented in excess with the supernatant of J558L plasmacytoma
cells stably transfected with a vector encoding murine IL7. Transformed cells were selected by IL7 withdrawal and kept in optimum conditions38. Retroviral vectors containing either
constitutively active STAT5 (STAT5-CA)34 or an empty vector (EV) were used to transduce BCR-ABL1-transformed cells and sorted cells were used then for western blot or flow cytometry
analysis. 1–2 µM 4-hydroxy Tam (Sigma-Aldrich) was used to induce deletion on plasmids expressing Tam-inducible form of Cre (Cre-ERT2)14. All cells were tested and found free from
mycoplasma. EXPRESSION ASSAYS Total RNA was isolated using the Direct-zol™ RNA Kit (Zymo Research) or ReliaPrep™ RNA Cell Miniprep System (Promega), and synthesis of cDNA was performed
(Thermo Fisher). Quantitative real time PCR analyses were performed on ABI7900HT PCR machine (Applied Biosystems) using Quantitect assays (Qiagen) and SYBR Green (Applied Biosystems). The
expression of _ABL1_ and the fusion _BCR-ABL_ (m-bcr; e1-a2) were measured using TaqMan Gene expression assays (Hs01104728_m1 ABL1 and Hs03024844_ft BCR-ABL, respectively) from Applied
Biosystems. Relative quantification was calculated using 2−ΔΔCT equation. RNA-SEQUENCING BM cells were isolated from two different mice and were then kept in culture with IL7 for 7 days.
Afterwards, pre-B cells were transduced with either an EV or with BCR-ABL1 retroviral vectors and kept for 48 h in +IL7 medium. Biological triplicates were prepared in three independent
experiments (_n_ = 6). Then, IL7 was removed from cells transduced with BCR-ABL1 for one week until cells were completely transformed. Pre-B cells transduced with EV were kept in culture
with IL7 for similar culturing time points as transformed cells, then sorted for GFP. Total RNA of pre-B cells transduced with either EV or with BCR-ABL1 was prepared using the ReliaPrep™
RNA Miniprep Kit (Promega). The total RNA library was generated using the Illumina TruSeq® stranded total RNA (Gold) kit and the multiplexed samples were sequenced on Illumina HiSeq 3000
machine to produce an average of ~100 million paired-end reads with 150 bp in length per sample. The base calling was performed by using BCL2Fastq pipeline (version: 0.3.0) and bcl2fastq
(version 2.17.1.14). PCA, Differential expression analysis and additional statistical tests related to RNA-seq were performed using R and bioconductor packages55,56. The broad MIT GSEA
application57 was used for GSEA. Detailed description of methods used in data analysis is provided in Supplementary Methods. IN SITU PROXIMITY LIGATION ASSAY (PLA) For PLA experiemtns25,58,
the cytokine IL7 and the chemokine CXCL12 were labeled with PLA-PLUS and PLA-MINUS probes. For PLA experiments with JAK3 or pJAK3 the corresponding antibodies were used (Cell Signaling). The
PLA probes were then subjected to ligation and polymerization reactions (Sigma-Aldrich). The cells were then examined for the frequency of signals per cell under the fluorescence microscope
(Leica). Pictures were taken and quantified Image J and BlobFinder software. IN VIVO TRANSPLANTATION OF MOUSE LEUKEMIA CELLS Mouse pre-B cells from _Il7r__α__fl/fl_ or _FoxO1__fl/fl_ were
transformed with pMIG-BCR-ABL1 (kindly provided by W. Pear) and contained either ERT2 or Cre-ERT2 were labeled with retroviral firefly luciferase and were then injected intravenously into
sublethally irradiated NOD-SCID mice38. Engraftment was monitored using luciferase bioimaging38. Mice were randomly allocated into each treatment group. XENOGRAFTS WITH HUMAN ALL SAMPLES
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were purchased from Charles River and bred. All mouse housing, breeding, and surgical procedures were approved by the governmental animal care and
use committees in Schleswig-Holstein (Ministerium für Energiewende, Landwirtschaft, Umwelt, Natur und Digitalisierung). 8–12-week-old female mice were injected intravenously with 1 × 106
ALL cells from patient BM (>90% blasts)59,60. Animals were sacrificed upon detection of >75% leukemic blasts or clinical leukemia (loss of weight or activity, organomegaly, and
hind-limb paralysis). Leukemia infiltration to spleen and BM was determined61. IMATINIB AND ANTIBODY TREATMENT IN VIVO NSG mice were injected with 1 × 106 BCR-ABL positive ALL cells/animal.
In total 40 mg/kg of imatinib (LC Laboratories) were administered orally 5 days a week. In total 1 mg/kg of anti-IL7Rα antibody (clone 40131, R&D Systems) or isotype control antibody
were injected intravenously on day +1, +3, +7, +21, and then every other week. Mice were sacrificed when they showed signs of leukemia or when they had at least 75% blasts in peripheral
blood. Mice were randomly allocated into each treatment group and no blinding was used. FLOW CYTOMETRY Antibodies for flow cytometry (CD19, IL7R, CXCR4, FOXO1, and CD11b) were purchased from
(eBioscience, BioLegend, Invitrogen, or Cell Signaling). Intracellular flow cytometry staining was performed using the Fix and Perm cell permeabilization kit (ADG). Cell viability was
measured using Sytox® blue dead cell stain (Life Technologies). FACS CantoII (BD Biosciences) was used for flow cytometry, and FlowJo v.10.1 was used for data analysis. More information
about the antibodies used in this study are provided in Supplementary Table 4. Detailed information for gating strategy is provided in Supplementary Fig. 14. MEASUREMENT OF CA2+ FLUX A total
of 1 × 106 cells were loaded with Indo-1 AM (Invitrogen) and used for Ca2+ analyses62. In total 100 ng/ml of CXCL12 was used for stimulation. WESTERN BLOT Wet-western blotting was
performed61. pJAK3, pJAK2, pJAK1, pSTAT5, pFOXO1, JAK1, JAK2, JAK3, STAT5, FOXO1, and GAPDH antibodies were obtained from Cell Signaling Technology. More information about the antibodies
used in this study are provided in Supplementary Table 4. All originals uncropped gels are provided in Supplementary Fig. 15. STATISTICS AND REPRODUCIBILITY Statistical tests are indicated
in the figure legends. Results were analyzed for statistical significance with GraphPad Prism 8.3.0 software or SPSS (v 24.0.0.2). A _p_ value of < 0.0500 was considered significant (*_p_
< 0.005, **_p_ < 0.001, ***_p_ < 0.001, ****_p_ < 0.001). In vitro panels are representative of at least three independent experiments, unless mentioned otherwise. REPORTING
SUMMARY Further information on research design is available in the Nature Research Reporting Summary linked to this article. DATA AVAILABILITY All data supporting the findings of this study
are available within the article and its supplementary information files. The sequencing data that support the findings in Fig. 1, Supplementary Figs 1 and 2, Supplementary Table 1, and
Supplementary Data 1 have been deposited in NCBI’s Gene Expression Omnibus63 and are accessible through GEO Series accession number GSE150784. A reporting summary for this article is
available as a Supplementary Information file. CODE AVAILABILITY The scripts used for analysis and figure generation are available at https://github.com/medhaniea/pca-and-heatmap. REFERENCES
* Ribeiro, R. C. et al. Clinical and biologic hallmarks of the Philadelphia chromosome in childhood acute lymphoblastic leukemia. _Blood_ 70, 948–953 (1987). Article CAS PubMed Google
Scholar * Arico, M. et al. Outcome of treatment in children with Philadelphia chromosome-positive acute lymphoblastic leukemia. _N. Engl. J. Med._ 342, 998–1006 (2000). Article CAS PubMed
Google Scholar * Schultz, K. R. et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children’s Oncology Group study
AALL0031. _Leukemia_ 28, 1467–1471 (2014). Article CAS PubMed PubMed Central Google Scholar * Alsadeq, A. et al. IL7R is associated with CNS infiltration and relapse in pediatric B-cell
precursor acute lymphoblastic leukemia. _Blood_ 132, 1614–1617 (2018). * Mazzucchelli, R. & Durum, S. K. Interleukin-7 receptor expression: intelligent design. _Nat. Rev. Immunol._ 7,
144–154 (2007). Article CAS PubMed Google Scholar * Park, L. S. et al. Cloning of the murine thymic stromal lymphopoietin (TSLP) receptor: Formation of a functional heteromeric complex
requires interleukin 7 receptor. _J. Exp. Med._ 192, 659–670 (2000). Article CAS PubMed PubMed Central Google Scholar * Geng, H. et al. Self-enforcing feedback activation between BCL6
and pre-B cell receptor signaling defines a distinct subtype of acute lymphoblastic leukemia. _Cancer Cell_ 27, 409–425 (2015). Article CAS PubMed PubMed Central Google Scholar *
Hertzberg, L. et al. Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the
International BFM Study Group. _Blood_ 115, 1006–1017 (2010). Article CAS PubMed Google Scholar * Sasson, S. C. et al. IL-7 receptor is expressed on adult pre-B-cell acute lymphoblastic
leukemia and other B-cell derived neoplasms and correlates with expression of proliferation and survival markers. _Cytokine_ 50, 58–68 (2010). Article CAS PubMed Google Scholar *
Mullighan, C. G. The molecular genetic makeup of acute lymphoblastic leukemia. _Hematology/the Education Program of the American Society of Hematology. American Society of Hematology_.
_Educ. Program_ 2012, 389–396 (2012). Google Scholar * Shochat, C. et al. Gain-of-function mutations in interleukin-7 receptor-alpha (IL7R) in childhood acute lymphoblastic leukemias. _J.
Exp. Med._ 208, 901–908 (2011). Article CAS PubMed PubMed Central Google Scholar * Kerdiles, Y. M. et al. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7
and interleukin 7 receptor. _Nat. Immunol._ 10, 176–184 (2009). Article CAS PubMed PubMed Central Google Scholar * Herzog, S., Reth, M. & Jumaa, H. Regulation of B-cell
proliferation and differentiation by pre-B-cell receptor signalling. _Nat. Rev. Immunol._ 9, 195–205 (2009). Article CAS PubMed Google Scholar * Herzog, S. et al. SLP-65 regulates
immunoglobulin light chain gene recombination through the PI(3)K-PKB-Foxo pathway. _Nat. Immunol._ 9, 623–631 (2008). Article CAS PubMed Google Scholar * Guo, F. et al. CXCL12/CXCR4: a
symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. _Oncogene_ 35, 816–826 (2016). Article CAS PubMed Google Scholar * van den Berk, L.
C. et al. Disturbed CXCR4/CXCL12 axis in paediatric precursor B-cell acute lymphoblastic leukaemia. _Br. J. Haematol._ 166, 240–249 (2014). Article PubMed CAS Google Scholar * Hayashi,
S. et al. Stepwise progression of B lineage differentiation supported by interleukin 7 and other stromal cell molecules. _J. Exp. Med._ 171, 1683–1695 (1990). Article CAS PubMed Google
Scholar * Nagasawa, T. CXCL12/SDF-1 and CXCR4. _Front. Immunol._ 6, 301 (2015). Article PubMed PubMed Central CAS Google Scholar * Haferlach, T. et al. Clinical utility of
microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. _J. Clin. Oncol._
28, 2529–2537 (2010). Article CAS PubMed PubMed Central Google Scholar * Li, J. F. et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an
international study of 1,223 cases. _Proc. Natl Acad. Sci. USA_ 115, E11711–E11720 (2018). Article ADS CAS PubMed PubMed Central Google Scholar * Jacobs, S. R., Michalek, R. D. &
Rathmell, J. C. IL-7 is essential for homeostatic control of T cell metabolism in vivo. _J. Immunol._ 184, 3461–3469 (2010). Article CAS PubMed Google Scholar * Peschon, J. J. et al.
Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. _J. Exp. Med._ 180, 1955–1960 (1994). Article CAS PubMed Google Scholar * Nie, Y. et al. The
role of CXCR4 in maintaining peripheral B cell compartments and humoral immunity. _J. Exp. Med._ 200, 1145–1156 (2004). Article CAS PubMed PubMed Central Google Scholar * Ptasznik, A.
et al. Crosstalk between BCR/ABL oncoprotein and CXCR4 signaling through a Src family kinase in human leukemia cells. _J. Exp. Med._ 196, 667–678 (2002). Article CAS PubMed PubMed Central
Google Scholar * Becker, M., Hobeika, E., Jumaa, H., Reth, M. & Maity, P. C. CXCR4 signaling and function require the expression of the IgD-class B-cell antigen receptor. _Proc. Natl
Acad. Sci. USA_ 114, 5231–5236 (2017). Article CAS PubMed PubMed Central Google Scholar * Shah, N. P. et al. Overriding imatinib resistance with a novel ABL kinase inhibitor. _Science_
305, 399–401 (2004). Article ADS CAS PubMed Google Scholar * Flemming, A., Brummer, T., Reth, M. & Jumaa, H. The adaptor protein SLP-65 acts as a tumor suppressor that limits pre-B
cell expansion. _Nat. Immunol._ 4, 38–43 (2003). Article CAS PubMed Google Scholar * Moriguchi, M. et al. CXCL12 signaling is independent of Jak2 and Jak3. _J. Biol. Chem._ 280,
17408–17414 (2005). Article CAS PubMed Google Scholar * Nosaka, T. & Kitamura, T. Janus kinases (JAKs) and signal transducers and activators of transcription (STATs) in hematopoietic
cells. _Int J. Hematol._ 71, 309–319 (2000). CAS PubMed Google Scholar * Goetz, C. A., Harmon, I. R., O’Neil, J. J., Burchill, M. A. & Farrar, M. A. STAT5 activation underlies IL7
receptor-dependent B cell development. _J. Immunol._ 172, 4770–4778 (2004). Article CAS PubMed Google Scholar * Dominguez-Sola, D. et al. The FOXO1 transcription factor instructs the
germinal center dark zone program. _Immunity_ 43, 1064–1074 (2015). Article CAS PubMed Google Scholar * Sander, S. et al. PI3 Kinase and FOXO1 transcription factor activity
differentially control B cells in the germinal center light and dark zones. _Immunity_ 43, 1075–1086 (2015). Article CAS PubMed Google Scholar * Santos, S. C. et al. Constitutively
active STAT5 variants induce growth and survival of hematopoietic cells through a PI 3-kinase/Akt dependent pathway. _Oncogene_ 20, 2080–2090 (2001). Article CAS PubMed Google Scholar *
Onishi, M. et al. Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. _Mol. Cell Biol._ 18, 3871–3879 (1998). Article CAS PubMed
PubMed Central Google Scholar * Frismantas, V. et al. Ex vivo drug response profiling detects recurrent sensitivity patterns in drug-resistant acute lymphoblastic leukemia. _Blood_ 129,
e26–e37 (2017). Article CAS PubMed PubMed Central Google Scholar * Baccarani, M. et al. Chronic myeloid leukemia: an update of concepts and management recommendations of European
LeukemiaNet. _J. Clin. Oncol._ 27, 6041–6051 (2009). Article CAS PubMed PubMed Central Google Scholar * Bradeen, H. A. et al. Comparison of imatinib mesylate, dasatinib (BMS-354825),
and nilotinib (AMN107) in an N-ethyl-N-nitrosourea (ENU)-based mutagenesis screen: high efficacy of drug combinations. _Blood_ 108, 2332–2338 (2006). Article CAS PubMed PubMed Central
Google Scholar * Shojaee, S. et al. PTEN opposes negative selection and enables oncogenic transformation of pre-B cells. _Nat. Med._ 22, 379–387 (2016). Article CAS PubMed PubMed Central
Google Scholar * Hixon, J.A., et al. New anti-IL-7Ralpha monoclonal antibodies show efficacy against T cell acute lymphoblastic leukemia in pre-clinical models. _Leukemia_ 34, 35–49
(2019). * Tummers, B. & Green, D. R. Caspase-8: regulating life and death. _Immunol. Rev._ 277, 76–89 (2017). Article CAS PubMed PubMed Central Google Scholar * Biondi, A. et al.
Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study.
_Lancet Oncol._ 13, 936–945 (2012). Article CAS PubMed PubMed Central Google Scholar * Druker, B. J. et al. Five-year follow-up of patients receiving imatinib for chronic myeloid
leukemia. _N. Engl. J. Med._ 355, 2408–2417 (2006). Article CAS PubMed Google Scholar * Ribeiro, D. et al. STAT5 is essential for IL-7-mediated viability, growth, and proliferation of
T-cell acute lymphoblastic leukemia cells. _Blood Adv._ 2, 2199–2213 (2018). Article PubMed PubMed Central Google Scholar * Ge, Z. et al. Co-existence of IL7R high and SH2B3 low
expression distinguishes a novel high-risk acute lymphoblastic leukemia with Ikaros dysfunction. _Oncotarget_ 7, 46014–46027 (2016). Article PubMed PubMed Central Google Scholar *
Mullighan, C. G. et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. _Nature_ 453, 110–114 (2008). Article ADS CAS PubMed Google Scholar * Churchman, M.
L. et al. Efficacy of retinoids in IKZF1-mutated BCR-ABL1 acute lymphoblastic leukemia. _Cancer Cell_ 28, 343–356 (2015). Article CAS PubMed PubMed Central Google Scholar * Appelmann,
I. et al. Janus kinase inhibition by ruxolitinib extends dasatinib- and dexamethasone-induced remissions in a mouse model of Ph+ ALL. _Blood_ 125, 1444–1451 (2015). Article CAS PubMed
PubMed Central Google Scholar * Singh, H. et al. A screening-based approach to circumvent tumor microenvironment-driven intrinsic resistance to BCR-ABL+ inhibitors in Ph+ acute
lymphoblastic leukemia. _J. Biomol. Screen_ 19, 158–167 (2014). Article CAS PubMed Google Scholar * Dorsey, J. F. et al. Interleukin-3 protects Bcr-Abl-transformed hematopoietic
progenitor cells from apoptosis induced by Bcr-Abl tyrosine kinase inhibitors. _Leukemia_ 16, 1589–1595 (2002). Article CAS PubMed Google Scholar * Roberts, K. G. et al. Targetable
kinase-activating lesions in Ph-like acute lymphoblastic leukemia. _N. Engl. J. Med._ 371, 1005–1015 (2014). Article PubMed PubMed Central CAS Google Scholar * von Freeden-Jeffry, U. et
al. Lymphopenia in interleukin (IL)−7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. _J. Exp. Med._ 181, 1519–1526 (1995). Article Google Scholar * Puel, A., Ziegler, S.
F., Buckley, R. H. & Leonard, W. J. Defective IL7R expression in T(−)B(+)NK(+) severe combined immunodeficiency. _Nat. Genet._ 20, 394–397 (1998). Article CAS PubMed Google Scholar *
Belarif, L. et al. IL-7 receptor blockade blunts antigen-specific memory T cell responses and chronic inflammation in primates. _Nat. Commun._ 9, 4483 (2018). Article ADS PubMed PubMed
Central CAS Google Scholar * Ellis, J. et al. Anti-IL-7 receptor alpha monoclonal antibody (GSK2618960) in healthy subjects—a randomized, double-blind, placebo-controlled study. _Br. J.
Clin. Pharmacol._ 85, 304–315 (2019). Article CAS PubMed Google Scholar * Huber, W. et al. Orchestrating high-throughput genomic analysis with bioconductor. _Nat. Methods_ 12, 115–121
(2015). Article CAS PubMed PubMed Central Google Scholar * R Core Team. R: a language and environment for statistical computing. (R Foundation for Statistical Computing, Vienna,
Austria, 2015). * Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. _Proc. Natl Acad. Sci. USA_ 102,
15545–15550 (2005). Article ADS CAS PubMed PubMed Central Google Scholar * Levit-Zerdoun, E. et al. Survival of igalpha-deficient mature B cells requires BAFF-R function. _J. Immunol._
196, 2348–2360 (2016). Article CAS PubMed Google Scholar * Krause, S. et al. Mer tyrosine kinase promotes the survival of t(1;19)-positive acute lymphoblastic leukemia (ALL) in the
central nervous system (CNS). _Blood_ 125, 820–830 (2015). Article CAS PubMed Google Scholar * Alsadeq, A. et al. Effects of p38alpha/beta inhibition on acute lymphoblastic leukemia
proliferation and survival in vivo. _Leukemia_ 29, 2307–2316 (2015). * Alsadeq, A. et al. The role of ZAP70 kinase in acute lymphoblastic leukemia infiltration into the central nervous
system. _Haematologica_ 102, 346–355 (2017). Article CAS PubMed PubMed Central Google Scholar * Kohler, F. et al. Autoreactive B cell receptors mimic autonomous pre-B cell receptor
signaling and induce proliferation of early B cells. _Immunity_ 29, 912–921 (2008). Article PubMed CAS Google Scholar * Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression
Omnibus: NCBI gene expression and hybridization array data repository. _Nucleic Acids Res._ 30, 207–210 (2002). Article CAS PubMed PubMed Central Google Scholar Download references
ACKNOWLEDGEMENTS This work was supported by the Deutsche Krebshilfe and Deutsche Forschungsgemeinschaft (SFB1074, projects A9 and A10; SFB1279, project B03) and the ERC advanced grant to
H.J. (694992). This work was also supported by the Chief Scientist Office grant to C.H. (ETM/374). H.A. is funded by the Egyptian Ministry of Higher Education (MoHE) and the German Academic
Exchange Service (DAAD) within the 6th call (2014–2015; GERLS: Section 441, No.91528030). H.A. is also funded by the Department of Molecular Biology, Genetic Engineering and Biotechnology
Division, National Research Centre (NRC) in Egypt. D.M.S. is supported by the Wilhelm Sander Stiftung (2016.110.1) and the Deutsche José-Carreras Leukämiestiftung (DJCLS 17R/2017). Research
in the M.M. laboratory is funded by the NIH/NCI through Outstanding Investigator Award R35CA197628, R01CA157644, R01CA213138, the Norman and Sadie Lee Foundation for Pediatric Cancer and the
California Institute for Regenerative Medicine (CIRM) through DISC2–10061. M.M. is a Howard Hughes Medical Institute (HHMI) Faculty Scholar. We thank Julia Lanzinger, Katrin Timm-Richert,
Katrin Neumann, Annette Tietz, and Gabriele Allies for the excellent technical assistance. We thank Sebastian Wiese and the core project of SFB1074. We thank Prof. Jinyan Huang for providing
RNA-Seq gene expression data for IL7R/CXCR4. AUTHOR INFORMATION Author notes * These authors contributed equally: Ameera Alsadeq, Hassan Jumaa. AUTHORS AND AFFILIATIONS * Institute of
Immunology, Ulm University Medical Center, 89081, Ulm, Germany Hend Abdelrasoul, Anila Vadakumchery, Markus Werner, Ahmad Khadour, Marc Young, Omar El Ayoubi, Markus Krämer, Vera Schmid,
Elias Hobeika, Ameera Alsadeq & Hassan Jumaa * Department of Pediatrics I, ALL-BFM Study Group, Christian-Albrechts University Kiel and University Medical Center Schleswig-Holstein,
Kiel, Germany Lennart Lenk, Fotini Vogiatzi, Gunnar Cario, Martin Schrappe & Denis M. Schewe * Department of Systems Biology and City of Hope Comprehensive Cancer Center, Monrovia, CA,
USA Zhengshan Chen & Markus Müschen * Institute of Cancer Sciences, College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow, UK Yasar Yousafzai & Christina
Halsey * Institute of Experimental Cancer Research, Medical Faculty, University of Ulm, Ulm, Germany Medhanie A. Mulaw Authors * Hend Abdelrasoul View author publications You can also search
for this author inPubMed Google Scholar * Anila Vadakumchery View author publications You can also search for this author inPubMed Google Scholar * Markus Werner View author publications
You can also search for this author inPubMed Google Scholar * Lennart Lenk View author publications You can also search for this author inPubMed Google Scholar * Ahmad Khadour View author
publications You can also search for this author inPubMed Google Scholar * Marc Young View author publications You can also search for this author inPubMed Google Scholar * Omar El Ayoubi
View author publications You can also search for this author inPubMed Google Scholar * Fotini Vogiatzi View author publications You can also search for this author inPubMed Google Scholar *
Markus Krämer View author publications You can also search for this author inPubMed Google Scholar * Vera Schmid View author publications You can also search for this author inPubMed Google
Scholar * Zhengshan Chen View author publications You can also search for this author inPubMed Google Scholar * Yasar Yousafzai View author publications You can also search for this author
inPubMed Google Scholar * Gunnar Cario View author publications You can also search for this author inPubMed Google Scholar * Martin Schrappe View author publications You can also search for
this author inPubMed Google Scholar * Markus Müschen View author publications You can also search for this author inPubMed Google Scholar * Christina Halsey View author publications You can
also search for this author inPubMed Google Scholar * Medhanie A. Mulaw View author publications You can also search for this author inPubMed Google Scholar * Denis M. Schewe View author
publications You can also search for this author inPubMed Google Scholar * Elias Hobeika View author publications You can also search for this author inPubMed Google Scholar * Ameera Alsadeq
View author publications You can also search for this author inPubMed Google Scholar * Hassan Jumaa View author publications You can also search for this author inPubMed Google Scholar
CONTRIBUTIONS H.A., A.A., A.V., M.W., A.K., and O.E. performed experiments in wildtype and BCR-ABL1-transformed murine cells. M.K. and V.S. performed PLA experiments. Z.C. and M.M performed
inducible IL7R and FOXO1 deletion in vivo. A.A., L.L., and F.V. performed experiments and analyzed xenograft models. Y.Y. performed experiments. M.Y. performed RNA-Seq analysis. M.A.M.
provided RNA-Seq data. E.H. provided PLA data. D.M.S., G.C., and M.S. provided ALL patient materials. A.A., E.H., D.M.S., and C.H. designed experiments and discussed the research direction.
A.A. prepared all figures and wrote paper. H.J. initiated, designed, supervised research, and wrote the paper. All authors discussed the paper. CORRESPONDING AUTHOR Correspondence to Hassan
Jumaa. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PEER REVIEW INFORMATION _Nature Communications_ thanks the anonymous
reviewers for their contribution to the peer review of this work. Peer reviewer reports are available. PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims
in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1
REPORTING SUMMARY RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation,
distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and
indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to
the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will
need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE
CITE THIS ARTICLE Abdelrasoul, H., Vadakumchery, A., Werner, M. _et al._ Synergism between IL7R and CXCR4 drives BCR-ABL induced transformation in Philadelphia chromosome-positive acute
lymphoblastic leukemia. _Nat Commun_ 11, 3194 (2020). https://doi.org/10.1038/s41467-020-16927-w Download citation * Received: 08 August 2019 * Accepted: 29 May 2020 * Published: 24 June
2020 * DOI: https://doi.org/10.1038/s41467-020-16927-w SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable
link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative