Depletion of loss-of-function germline mutations in centenarians reveals longevity genes
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT While previous studies identified common genetic variants associated with longevity in centenarians, the role of the rare loss-of-function (LOF) mutation burden remains largely
unexplored. Here, we investigated the burden of rare LOF mutations in Ashkenazi Jewish individuals from the Longevity Genes Project and LonGenity study cohorts using whole-exome sequencing
data. We found that centenarians had a significantly lower burden (11-22%) of LOF mutations compared to controls. Similar effects were also observed in their offspring. Gene-level burden
analysis identified 35 genes with depleted LOF mutations in centenarians, with 14 of these validated in the UK Biobank. Mendelian randomization and multi-omic analyses on these genes
identified _RGP1_, _PCNX2_, and _ANO9_ as longevity genes with consistent causal effects on multiple aging-related traits and altered expression during aging. Our findings suggest that a
protective genetic background, characterized by a reduced burden of damaging variants, contributes to exceptional longevity, likely acting in concert with specific protective variants to
promote healthy aging. SIMILAR CONTENT BEING VIEWED BY OTHERS RARE GENETIC CODING VARIANTS ASSOCIATED WITH HUMAN LONGEVITY AND PROTECTION AGAINST AGE-RELATED DISEASES Article 13 September
2021 THE 90 PLUS: LONGEVITY AND COVID-19 SURVIVAL Article 08 February 2022 THE BURDEN OF RARE PROTEIN-TRUNCATING GENETIC VARIANTS ON HUMAN LIFESPAN Article Open access 03 March 2022
INTRODUCTION Aging is a complex process characterized by an accumulation of molecular damage, progressive decline in physiological function, increased susceptibility to disease, and,
ultimately, higher risk of mortality1. While chronological age is a major risk factor, there is remarkable variability in how individuals age, with some experiencing severe disability and
premature death while others maintain good health well into old age2,3. This heterogeneity suggests that aging is a multifactorial process shaped by both genetic and environmental factors4.
At the extreme end of the lifespan spectrum are centenarians, individuals with exceptional longevity who have reached the age of 100 years or more. Centenarians represent a rare and valuable
model of successful aging, often displaying delayed onset or escape from major age-related diseases such as cardiovascular disease, diabetes, and dementia5,6. Furthermore, many maintain
physical and cognitive function, as well as independence, well into old age7. Understanding the factors that contribute to their exceptional longevity could provide valuable insights into
the biology of healthy aging and lifespan determination. Studies in model organisms have firmly established that lifespan has a significant genetic component. Single gene mutations in
pathways related to insulin/insulin-like growth factor-1 (IGF-1) signaling, mechanistic target of rapamycin (mTOR) signaling, and AMP-activated protein kinase (AMPK) signaling have been
shown to dramatically extend lifespan in yeast, worms, flies, and mice8. Many of these pathways are evolutionarily conserved, suggesting they may play a role in human aging as well. Indeed,
functional variants in the IGF-1 receptor have been identified in centenarians, supporting a role for this pathway in exceptional longevity9. In humans, genome-wide association studies
(GWAS) have identified numerous common genetic variants associated with longevity, defined as attaining exceptional old age or having long-lived parents10,11. However, these variants explain
only a small portion of the heritability (12%)11, suggesting that rare variants may also play an important role12. Rare variants, particularly those that lead to loss of gene function
(LOF), are of great interest in studying human lifespan. LOF variants, including nonsense, splice-site, and frameshift mutations, are generally deleterious and subject to strong purifying
selection13. An increased burden of LOF mutations has been observed in individuals with shorter lifespans and shorter period of life people spent free of disease (or healthspan), suggesting
this may significantly impact human health14. However, LOF variants that confer protective effects, such as those in the _APOC3_ and _PCSK9_ genes associated with a lower risk of
cardiovascular disease have also been identified15,16. Despite the growing evidence for the importance of rare variants in aging, the overall burden of LOF mutations in exceptionally
long-lived individuals compared to controls has not been systematically examined. A previous study observed no difference in the burden of pathogenic variants between centenarians, their
offspring, and controls17. However, this study did not specifically focus on LOF variants or incorporate key covariates that may introduce batch effects confounding the results. Furthermore,
the sample size was smaller than in the present study, limiting the power to detect significant differences. Another study found that the burden of rarest protein-truncating variants (PTVs)
in two large cohorts was negatively associated with human healthspan and lifespan, accounting for 0.4 and 1.3 years of their variability, respectively14. In this study, we leveraged
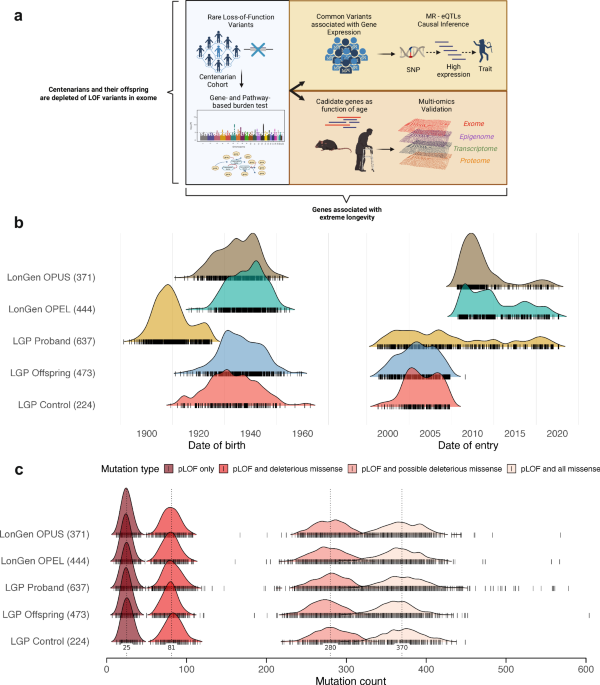
whole-exome sequencing data from a large cohort of Ashkenazi Jewish centenarians and controls to comprehensively compare the burden of rare LOF variants (Fig. 1a). By focusing on a
genetically homogeneous population, we minimized the potential confounding effects of population stratification. Importantly, we incorporated the dates of recruitment and birth as
coefficients in our analysis to control for cohort effects and potential secular trends in environmental and lifestyle factors that may impact lifespan. Our results suggest that centenarians
have a lower burden of LOF mutations compared to controls. This depletion was observed across multiple categories of predicted deleterious variants. Furthermore, we performed a genome-wide
association study to identify specific genes and pathways that were enriched for protective variants in centenarians. Several genes reached suggestive significance levels, and pathway
analysis revealed a depletion of variants in pathways related to hyaluronan metabolism, G-protein receptors, post-translational protein modification, and mitochondrial translation. Notably,
14 out of 35 of these gene associations were validated in an independent cohort from the UK Biobank based on parental lifespan-related traits, supporting the reproducibility of our findings.
Together, these results provide new insights into the genetic architecture of human exceptional longevity and highlight potential molecular mechanisms that may contribute to healthy aging.
Further studies will be necessary to validate and functionally characterize the roles of these genes and pathways in promoting longevity. RESULTS The whole-exome sequencing data was obtained
from 637 centenarians, 917 offspring of centenarians, and 595 controls from the Longevity Genes Project (LGP) and LonGenity study cohorts of Ashkenazi Jewish individuals (Table 1, Fig. 1a,
Methods)18. Based on the demographic characteristics, participants were recruited continuously over a period of 20 years (2000–2020). However, the recruited centenarians were mostly born
between 1900 and 1920, while most of the offspring and controls were born between 1920 and 1960 (Fig. 1b). This suggests that the direct comparison of mutation burden between centenarians
and controls may potentially be confounded by the date of recruitment and date of birth. We identified loss-of-function (LOF) mutations based on the following criteria: alternate allele
frequency (AAF) < 1%, Hardy–Weinberg equilibrium (HWE) threshold of 10−15, and variant missingness <10%. We classified the variants into different categories based on their predicted
deleteriousness: pLOF only, pLOF and missense, pLOF and predicted deleterious missense (5/5 algorithms predict a deleterious variant), and pLOF and predicted deleterious missense (at least
1/5 algorithms predict a deleterious variant). The deleteriousness of missense variants was assessed using five different computational methods (Method). We counted the cumulative mutation
burden in centenarians, their offspring, and controls across different categories of predicted deleterious variants. Consistent with the potential confounding effects of dates of recruitment
and birth, we initially observed a similar distribution of LOF mutations across the different categories in centenarians and controls (Fig. 1c). We performed quality control and filtering,
retaining 338 centenarians with recorded age over 100 years old and 420 controls with age less than 90 years old (Table 1, Methods). We observed a similar distribution of raw mutation count
after filtering (Supplementary Fig. 1) We then performed the count-based burden test using linear regression models. Furthermore, we found that even without adjusting for potential
confounders, offspring but not centenarians showed a significantly lower mutation burden in all pLOF categories (Supplementary Fig. 2). This is likely due to the smaller batch effect between
offspring group and control, compared to the centenarian group. We also showed that there is no significant difference observed between centenarians and their offspring (Supplementary Fig.
3). To account for these potential confounders, we binned the dates of recruitment and birth and added them as coefficients in the burden test model. After adjusting for these covariates, we
found a consistent and significant trend of lower burden of LOF mutations in centenarians and their offspring compared to controls across all categories of predicted deleterious variants
(Fig. 2). Notably, the depletion of LOF variants was statistically significant for centenarians in all categories, including the pLOF-only category (_b_ = −5.5, _p_ = 0.0453). The effect
sizes for centenarians ranged from −5.5 to −39.6, indicating a 11% to 22% reduction in mutation burden compared to controls. Furthermore, the offspring of centenarians also exhibited a
significantly lower mutation burden compared to controls in both the LGP (related to LGP centenarians) and LonGenity (unrelated to LGP centenarians) cohorts (Fig. 2). The effect sizes for
offspring were smaller than those observed for centenarians, but still significant, with _p_-values ranging from 1.17e-07 to 4.99e-4 in the LGP cohort and from 4.52e-4 to 0.021 in the
LonGenity cohort. These results suggest that the protective effect of a lower LOF mutation burden may be inherited by the offspring of centenarians, contributing to their increased
likelihood of exceptional longevity. We also performed a sensitivity analysis by using different covariates, including age at recruitment, top 10 genetic principal components, numerical date
of birth, and date of recruitment, and found consistent results for centenarian offspring in the LGP cohort (Supplementary Fig. 4). Statistical significance for centenarians and the
LonGenity cohort was sensitive to the choice of covariates, suggesting that the genetic associations with longevity are complex and possibly influenced by unmeasured factors. To identify
specific genes and pathways that carry a lower mutation burden in centenarians, we performed a gene-level and pathway-level burden test. The gene-level analysis identified 35 genes that
reached the significance level at FDR < 0.05 (Fig. 3a). Remarkably, 14 out of these 35 genes were validated in an independent study from the UK Biobank using parental lifespan-related
traits (Fig. 3a)19. Note that this is an indirect validation as the genetics of exceptional longevity and parental lifespan, while having similarities, may still obtain different
characteristics. Pathway-level analysis revealed processes related to hyaluronan metabolism, Class A/1 (Rhodopsin-like receptors), post-translational protein modification, and mitochondrial
translation reached the significance level at FDR < 0.05 (Fig. 3b). We observed a mild inflation in our test statistics, with a genomic inflation factor (λ) of 1.57. After adjusting for
the inflation, the top three pathways still reached the suggestive FDR threshold of 0.2. These results suggest that the depletion of mutations in these pathways may contribute to exceptional
longevity. To further investigate the potential causal effects of the identified longevity-associated genes on lifespan-related traits, we performed Mendelian Randomization (MR) analyzes
using public blood gene expression QTL data from eQTLgen and GWAS summary statistics of multiple lifespan-related traits (Fig. 4a)20. It is important to note that while the MR analysis uses
common variants (eQTLs) rather than rare coding variants, it can provide complementary evidence about a gene’s role in longevity through different mechanisms. MR analysis revealed that seven
genes had significant causal effects on multiple lifespan-related traits, such as frailty index, healthspan, lifespan, and extreme longevity (90th and 99th percentiles), and lifespan-GIP1
(the genetic principal component of healthy longevity, Methods). Among them, three genes (_RGP1_, _PCNX2_, and _ANO9_) showed consistent pro-longevity effects across the multiple traits
tested, supporting their potential roles in promoting longevity as suggested by burden analysis, while the other four genes showed anti-longevity effects. On the other hand, two of the genes
(_DYNC1H1_ and _GALNT12_) only show a significant protective effect on one trait (lifespan and extreme longevity at 99th percentile), while _PKP4_ only shows a significant positive effect
on healthspan but not in other traits. The other four genes (_ZNF446_, _PLA2G4B_, _EFNA3_, and _ABCF3_) show inconsistent effects on lifespan-related traits. We then profiled the multi-omic
associations of the identified longevity-associated genes to provide a systematic evaluation of their expression and regulation during aging (Fig. 4b–e). Comparison with exome-wide
gene-level associations with parental lifespan obtained from GeneBass (Fig. 4b)19, showed that six out of seven causal genes were significantly associated with parental lifespan, three genes
(_MLXIP_, _PCNX2_, and _DYNC1H1_) remain significant after corrected with multiple-testing with FDR. Analysis of age-related changes in promoter DNA methylation using data from 500
individuals in the Massachusetts General Brigham (MGB) biobank (Fig. 4c) revealed significant changes for most longevity-associated genes, except two (_RGP1_ and _BCLAF1_). Similarly,
age-related changes in blood gene expression obtained from the transcriptome-wide association study (TWAS) for aging by Peters et al. (Fig. 4d) showed significant changes for genes such as
_OPN3_, _PCNX2_, _GALNT12_, and _RGP1_21. Furthermore, age-related changes in plasma protein levels using Olink data from 53,015 UK Biobank participants (Fig. 4e) revealed significant
changes for proteins encoded by _DYNC1H1_ and _FLT4_ genes. The results suggest that the expression and regulation of these longevity-associated genes are altered during the aging process.
To gain further insights into the potential relevance of the identified longevity-associated genes in aging and interventions, we further compared their significance scores across different
signatures of aging and longevity interventions (Fig. 4f)22,23. The signature analysis results in 69 significant associations after adjusting for multiple testing of 266 tests using FDR
(Fig. 4f). It revealed that many of these genes (18 out of 21 tested) were also significantly associated with aging in humans and rodents, as well as with interventions known to extend
lifespan, such as caloric restriction (_ABCF3_, _CKAP2L_, and _CEP68_), rapamycin treatment (_PKP4_, _CTNND1_, and _RTRAF_), growth hormone deficiency (_HOGA1_, _ANKRD33_, and _MLXIP_), as
well as overall lifespan after intervention (_HOGA1_). Together, this multi-layered evidence supports the potential roles of these genes in regulating healthy aging and longevity. DISCUSSION
In this study, we have discovered that centenarians, within the large cohort we examined, possess a significantly lower burden of predicted deleterious LOF variants compared to controls.
This finding suggests that a protective genetic background, characterized by the depletion of damaging coding mutations, contributes to the exceptional longevity of centenarians. Notably, we
also observed a lower mutation burden in centenarian offspring, although the effect was less pronounced. These findings support the notion of a heritable component to longevity outside of
protective and common variants and suggest that the combined genetic background, including protective variants and depletion of damaging variants, may be transmitted across generations to
support exceptional longevity. Our results are consistent with previous studies that reported an increased burden of LOF variants in individuals with shorter lifespans and age-related
diseases14,24, and provide further evidence for the role of rare coding variants in extreme human longevity. Our study extends these findings by demonstrating that the depletion of LOF
variants in centenarians is not limited to the rarest variants but is observed across multiple categories of predicted deleterious variants. However, our findings contrast with those of
another study that observed no difference in the burden of pathogenic variants between centenarians, their offspring, and controls17. This discrepancy may be due to differences in study
design, such as the focus on LOF variants specifically, the larger sample size of our study, and the adjustment for potential confounding factors such as date of recruitment, age at
recruitment, and date of birth. Besides, due to the retrospective nature of the centenarian study, the centenarians usually have different demographic properties (age, date of birth, and
potentially other early life exposures) compared to the control group. While this can be addressed by including these features as covariates, this demographic disparity between centenarians
and controls emerges as a critical factor limiting the statistical power of centenarian studies (Fig. 1). In contrast, centenarian offspring, demographically more similar to controls, yield
stronger statistical evidence, corroborating our findings in centenarians. Future prospective studies with improved demographic matching are essential to elucidate the role of LOF variants
in exceptional longevity. Our pathway analysis revealed that centenarian exomes are depleted of LOF variants in several pathways related to aging and disease, including Class A/1
(Rhodopsin-like receptors), hyaluronan metabolism, post-translational protein modification, and mitochondrial translation. Class A/1 (Rhodopsin-like) receptors are involved in various
physiological processes and have been implicated in age-related diseases, suggesting their potential role in longevity25. Hyaluronan is a key component of the extracellular matrix that has
been shown to decline with age, and its increase contributes to the extension of lifespan26. Variants that maintain hyaluronan homeostasis may, therefore, promote healthy aging in humans.
Post-translational protein modifications play crucial roles in protein function and stability, and their dysregulation has been associated with various age-related diseases1. Mitochondrial
translation has also been linked to lifespan extension in model organisms27. To complement our analysis of rare LOF variants, we also investigated the causal role of identified longevity
genes in aging-related traits using MR analyzes. This approach allows us to infer potential causal relationships between gene expression and phenotypes of interest by using eQTLs (common
variants that are associated with gene expression) as instrumental variables. Our MR analyzes provided evidence for the causal effects of several longevity-associated genes, including
_RGP1_, _PCNX2_, and _ANO9_, on multiple aging-related traits. _PCNX2_ was identified to be associated with longevity in an independent GWAS study28, while _ANO9_ was associated with various
cancers29. These findings suggest that these genes may directly influence the aging process and contribute to the extended healthspan and lifespan. The consistent causal effect estimates
across different aging-related traits further support the robustness of these associations. Interestingly, our analyzes also revealed genes with more nuanced effects on longevity. For
instance, _DYNC1H1_ and _GALNT12_ showed significant deleterious effects on only one trait each (lifespan and extreme longevity at the 99th percentile, respectively), while _PKP4_
demonstrated a significant positive effect solely on healthspan. This suggests that these genes may influence particular aspects of the aging process rather than having a broad impact on all
longevity-related traits. Moreover, the inconsistent effects observed for genes such as _ZNF446_, _PLA2G4B_, _EFNA3_, and _ABCF3_ across different lifespan-related traits underscore the
complexity of genetic influences on aging and longevity. The multi-omic analyzes revealed that the expression and regulation of many longevity-associated genes are altered during aging,
specifically, 29 out of 31 for DNA methylation, 4 out of 11 for gene expression, and 2 out of 2 for plasma protein (Fig. 4). Follow-up studies are needed to elucidate the specific mechanisms
by which these genes and their encoded proteins contribute to healthy aging and longevity. Future studies could also explore the relationship between the burden of deleterious germline
mutations and the rate of biological aging in centenarians and the general population. Epigenetic clocks, which measure biological age based on DNA methylation patterns, have emerged as a
promising tool for assessing the pace of aging30,31. Previous studies have shown that centenarians exhibit slower epigenetic aging rates compared to the general population32. Integrating
rare variant burden data with epigenetic clock measures could provide novel insights into the interplay between genetic and epigenetic factors in shaping the rate of aging and exceptional
longevity, especially with current standardized tools like ClockBase and Biolearn33,34, as well as advanced aging clocks, including GrimAge235, DunedinPace36, and causality-enriched
clocks37. Such studies may uncover whether the reduced burden of harmful mutations observed in centenarians contributes to their slower biological aging rates. Our study also has several
limitations. First, while we adjusted for several important covariates, there may be other confounding factors that were not accounted for, such as environmental exposures and lifestyle
factors. Second, our study focused on a specific population (Ashkenazi Jews), although validation analysis in UK biobank suggests that the result may be generalizable to other ethnic groups.
Future studies in diverse populations will be necessary to confirm the generalizability of our findings. Third, the validation analysis is based on parental lifespan traits in the UK
biobank. Although previous studies on common variants show a substantial similarity between parental lifespan and exceptional longevity (rg = 0.81)38, it is unclear how similar the rare
genetic variants contribute to these two traits. Future validation and meta-analysis with other centenarian cohorts may help strengthen the robustness of our findings. Fourth, our study
relied on computational predictions of variant deleteriousness, which may not always reflect the true biological impact of a variant. Functional studies will be necessary to validate the
causal roles of the identified variants and genes in longevity. It is important to acknowledge that some LOF and missense variants can be protective, as demonstrated by previous
studies39,40,41. However, our hypothesis is that the overall probability of LOF variants being protective is lower than the probability of them being deleterious. This is because damaging a
component in a complex system is more likely to have a detrimental effect than a protective one42. Additionally, there is a selection bias, as highly damaging mutations are under-represented
in the population, while highly protective mutations are preserved43. These factors may explain the small effect sizes observed in our study. It should also be noted that we did not
identify any protective LOF variants (i.e., enrichment of LOF variants in centenarians) as demonstrated in previous study18, because we used a one-tailed test, focusing only on the depletion
of LOF variants. In conclusion, our study provides new insights into the genetic architecture of human exceptional longevity, exemplified by individuals who live to 100 years or beyond,
highlighting the importance of rare LOF variants and identifying novel genes and pathways that may promote healthy aging. We demonstrate that centenarians have a lower burden of predicted
deleterious LOF variants compared to controls and that this protective genetic background may be transmitted across generations. Our findings also underscore the complex interplay between
genetic variation, environmental factors, and age-related diseases in shaping human lifespan. Further studies in diverse populations and integrating multiple omics data will be necessary to
fully elucidate the mechanisms underlying exceptional longevity and develop targeted interventions to promote healthy aging. Nonetheless, our results represent an important step towards
understanding the genetic basis of human longevity and provide a foundation for future studies in this field. METHODS STUDY POPULATION AND DATA COLLECTION The study population was derived
from two ongoing studies of aging and longevity in the Ashkenazi Jewish population: the cross-sectional Longevity Genes Project (LGP) and the longitudinal LonGenity study18. The LGP cohort
consisted of 637 individuals with exceptional longevity, 473 offspring of long-lived individuals, and 224 controls, while the LonGenity cohort included 444 offspring of centenarians and 371
controls. All participants provided written informed consent, and the study was approved by the Institutional Review Board at Albert Einstein College of Medicine. For the analysis, we
applied filtering criteria to ensure the inclusion of appropriate individuals in each group. In the centenarian group, we removed individuals with a death or dropout record before 100
years, retaining 338 exceptionally long-lived centenarians. Similarly, in the control group, we removed individuals without death or dropout record before 90 years, resulting in 147
individuals from the LGP cohort and 273 individuals from the LonGenity cohort being included in the analysis (Table 1). WHOLE-EXOME SEQUENCING DNA samples from all participants were
subjected to whole-exome sequencing using the Illumina HiSeq 2000 platform at the Regeneron Genetics Center17. The sequencing reads were aligned to the human reference genome (hg38) using
the Burrows-Wheeler Aligner (BWA-mem v0.7.17)44, and duplicate reads were removed using Picard tools (version 1.96, http://broadinstitute.github.io/picard/). Variant calling was performed
using the Genome Analysis Toolkit (GATK v3.7)45. QUALITY CONTROL AND VARIANT ANNOTATION After genomic principal component analysis (PCA), four individuals with non-European ancestry were
excluded from the study. Quality control filtering was applied to remove potentially false-positive variants and genotype calls. Variants were filtered based on the following criteria:
alternate allele frequency (AAF) < 1% in the Ashkenazi Jewish population, Hardy–Weinberg equilibrium (HWE) _P_-value > 10−15, and variant missingness <10%, as suggested by a
previous study46. After QC filtering, autosomal-only variants with a minimum allele count (MAC) of 1 were divided into sets for centenarians, offspring, and controls for downstream analysis.
Loss-of-function (LOF) variants were defined as nonsense, splice-site, or frameshift mutations. Missense variants were classified as (1) possible deleterious missense mutation if they were
predicted to be damaging by at least 1 out of 5 algorithms (SIFT47, Polyphen2_HDIV48, Polyphen2_HVAR48, LRT49, and MutationTaster50) or (2) deleterious missense mutation if all five
algorithms predicted them to be damaging. SIFT (v6.2.1), Polyphen2_HDIV (v2.2.2), Polyphen2_HVAR (v2.2.2), LRT (v2016), and MutationTaster (v2021) were used in this analysis. BURDEN TEST
ANALYSIS Prior to the burden test, we removed the individual in the extreme longevity group with a lifespan or last reported age less than 100 years old. Therefore, only the 338 centenarians
are kept. Similarly, individuals in the control group with last reported age larger than 90 years old were also removed, with the remaining 147 individuals from LGP and 273 individuals for
lonGenity (Table 1). Descriptive statistics were used to summarize the demographic characteristics of the study population. The cumulative mutation burden for each individual was calculated
as the total number of population-level LOF (pLOF) and predicted deleterious missense variants. Mutation burden is calculated based on different categories of predicted deleterious variants
(pLOF only, pLOF and deleterious missense [5/5 algorithms], and pLOF and possible deleterious missense [≥1/5 algorithms], pLOF and all missense). Count-based burden tests were performed
using linear models with binned covariates to account for potential confounding factors, such as date of recruitment, date of birth, gender, age at visit, and top four genomic principal
components51. The cumulative mutation burden was used as the dependent variable, and the independent variables included centenarian status (or offspring status), binned date of recruitment,
and binned date of birth. Sensitivity analyzes were conducted by including additional covariates, such as age at recruitment, top 10 genetic principal components, numerical date of birth
(i.e., number of days since 1900-01-01), and date of recruitment. GENE-LEVEL AND PATHWAY-LEVEL BURDEN ANALYSIS Gene-level and pathway-level burden tests were performed using linear models,
with the cumulative mutation burden in each gene or pathway as the dependent variable and centenarian status as the independent variable. Only genes containing at least five pLOF variants
across the cohort were included. In total, 4925 unique genes were tested, and the significance threshold for gene-level tests was set at FDR < 0.05. Significant gene-level associations
were replicated using summary statistics from a gene-based association study of paternal or maternal lifespan in the GeneBass from UK biobank19. The significance threshold for replication
was set at _P_ < 0.05. MENDELIAN RANDOMIZATION To investigate the causal relationships between gene expression and aging-related traits, we performed Mendelian Randomization (MR) analyzes
using blood cis-eQTL data from eQTLgen, which includes 31,684 blood samples from 37 studies20. The outcome traits included aging-GIP1, frailty index, healthspan, lifespan, and extreme
longevity (90th and 99th percentiles). The parental lifespan GWAS was used as a proxy for individual lifespan and included 512,047 mothers and 500,193 fathers of European ancestry11. The
extreme longevity GWAS included 11,262 European subjects with a lifespan above the 90th percentile and 25,483 controls below the 60th percentile age10. Healthspan, defined as the age of the
first incidence of major age-related diseases or death, was analyzed using a GWAS of 300,447 UK Biobank participants aged 37–7352. The frailty index GWAS included 164,610 UK Biobank
participants aged 60–70 and 10,616 Swedish TwinGene participants aged 41–8753. Aging-GIP1, the first genetic principal component of six human aging traits, captures both length of life and
well-being indices54. We performed cis-Mendelian Randomization following the approach described by Ying et al37. Genetic variants strongly associated with whole blood gene expression levels
(FDR < 0.05) were selected as instrumental variables for the MR analysis. To minimize pleiotropic effects, only cis-eQTLs (located within 2 MB of target genes) were used, and LD clumping
was applied to remove eQTLs with strong LD (_r_2 > 0.3). We employed three MR methods based on the number of available eQTLs: Wald ratio for a single eQTL, generalized inverse variance
weighted (gIVW) for at least two eQTLs, and generalized MR-Egger regression (gEgger) for at least three eQTLs55. The gEgger method is robust to directional pleiotropy, we therefore reported
the P value from gEgger if pleiotropy is detected by gEgger intercept. MULTI-OMIC ANALYSIS OF THE IDENTIFIED LONGEVITY-ASSOCIATED GENES To systematically evaluate the expression and
regulation of the identified longevity-associated genes during aging, we profiled their multi-omic associations using various datasets. We obtained the exome-wide gene association with
parental lifespan using summary statistics from GeneBass19. Blood gene expression changes with age were obtained from the transcriptome-wide association study (TWAS) for aging by Peters et
al21. Age-related changes in promoter DNA methylation were assessed using data from 500 individuals in the Mass General Brigham (MGB) Biobank, which is also described in this study56. DNA
methylation profiles were generated using the Illumina Infinium MethylationEPIC v2.0 array, which covers over 935,000 CpG sites enriched for regulatory regions56. The cohort comprised
subjects of diverse ages, roughly balanced between male and female, and generally representative of the racial/ethnic distribution of the local area. For each CpG site associated with our
identified longevity-associated genes, we performed a linear regression to predict the methylation beta value using age, where the regression coefficient and _p_-value are calculated. The
CpG with the strongest association with age is used to represent the result. Age-related changes in plasma protein levels were investigated using Olink proteomics data from 53,015 UK Biobank
participants (UK Biobank Record Table 1072). Only two of our identified longevity-associated genes are presented in the Olink panel. We then performed a linear regression to predict the
protein level using age, where the regression coefficient and _p_-value are calculated. FDR was applied to adjust for multiple testing of all 471 sites tested. We performed FDR to adjust for
multiple tests in each omic layer. LONGEVITY SIGNATURE ANALYSIS To further explore the potential relevance of the identified longevity-associated genes in aging and interventions, we
compared their significance scores across different signatures of aging and longevity interventions using the GENtervention database57. For transcriptomic signatures of lifespan-extending
interventions, we selected the ones reflecting the most established longevity interventions that were identified based on gene expression data from at least 3 independent sources, as
described in Tyshkovskiy et al. 201923. The signatures included human aging and rodent aging, and interventions (caloric restriction, rapamycin treatment, and growth hormone deficiency). We
also include signatures of lifespan across interventions based on a larger set of longevity and lifespan-shortening interventions22. The significance scores were calculated as the
-log10(_P_-value) multiplied by the sign of the effect size (beta) for each gene in each signature. Nominal significance was set at _P_ < 0.05. Hierarchical clustering with Euclidean
distance was performed for the genes based on significance score. STATISTICS & REPRODUCIBILITY The study included a total of 2149 participants: 338 centenarians (aged 100 or older), 917
offspring of long-lived individuals, and 894 controls. Detailed age and sex/gender breakdowns for each group are provided in Table 1. Sex and gender were considered in the study design and
determined based on self-report at the time of recruitment. All participants provided written informed consent as stated in the “Study population and data collection” section. Participants
were not compensated for their involvement in the study. No statistical method was used to predetermine the sample size. Data exclusion criteria are detailed in the “Study population and
data collection” section. No other data were excluded from the analyzes. Statistical analyzes primarily employed linear models for burden tests and Mendelian Randomization, with adjustments
for potential confounding factors as described in the “Burden test analysis” and “Mendelian Randomization” sections. Multiple testing corrections were applied using FDR. The experiments were
not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment, as this was an observational genetic study. Reproducibility was addressed
through replication in independent datasets (UK Biobank). REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this
article. DATA AVAILABILITY All summary statistics for the gene- and pathway-based burden tests in the Ashkenazi Jewish longevity cohort are available in Supplementary Data 1, Supplementary
Data 2, and Source Data files. The individual-level genetic data from the Einstein longevity study are available under restricted access due to privacy concerns of research participants.
Qualified academic investigators (typically faculty members or postdoctoral researchers with relevant expertize) can request access by contacting Dr. Nir Barzilai
([email protected]) and the study’s principal investigator, Dr. Vadim Gladyshev ([email protected]). We aim to respond to all requests within 10 business days.
Access is subject to approval by the Institutional Review Board and requires a material transfer agreement. Upon approval, data use will be restricted by a comprehensive data use agreement
that includes conditions such as using the data solely for the approved research purpose, maintaining participant anonymity, and acknowledging the Einstein longevity study in any resulting
publications. Exact procedures for data transfer will be provided upon approval. The UK Biobank data used for validation is available through application to the UK Biobank
(https://www.ukbiobank.ac.uk/). Summary statistics from the eQTLGen consortium are publicly available at https://www.eqtlgen.org/. The GeneBass exome-wide association results are publicly
accessible at https://genebass.org/. Other publicly available datasets used in this study include: parental lifespan GWAS summary statistics (https://datashare.ed.ac.uk/handle/10283/3209),
healthspan GWAS summary statistics (https://www.gwasarchive.org/), frailty index GWAS summary statistics
(https://figshare.com/articles/dataset/Genome-Wide_Association_Study_of_the_Frailty_Index_-_Atkins_et_al_2019/9204998), longevity GWAS summary statistics
(https://www.longevitygenomics.org/downloads/). Source data are provided with this paper. CODE AVAILABILITY All of the analyzes are done in R 4.1. The custom code used for the burden test
analysis and gene-level and pathway-level burden analysis is available at https://doi.org/10.5281/zenodo.13756349 with a detailed readme file58. Other software used in our analysis was open
source and is described in the Methods section of the manuscript. REFERENCES * López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. _Cell_ 153,
1194–1217 (2013). Article PubMed PubMed Central Google Scholar * Beard, J. R. et al. The World report on ageing and health: a policy framework for healthy ageing. _Lancet_ 387,
2145–2154 (2016). Article PubMed Google Scholar * Lowsky, D. J., Olshansky, S. J., Bhattacharya, J. & Goldman, D. P. Heterogeneity in healthy aging. _J. Gerontol. A. Biol. Sci. Med.
Sci._ 69, 640–649 (2014). Article PubMed Google Scholar * Melzer, D., Pilling, L. C. & Ferrucci, L. The genetics of human ageing. _Nat. Rev. Genet._ 21, 88–101 (2020). Article CAS
PubMed Google Scholar * Andersen, S. L., Sebastiani, P., Dworkis, D. A., Feldman, L. & Perls, T. T. Health span approximates life span among many supercentenarians: compression of
morbidity at the approximate limit of life span. _J. Gerontol. A. Biol. Sci. Med. Sci._ 67A, 395–405 (2012). Article PubMed Central Google Scholar * Milman, S. & Barzilai, N.
Discovering biological mechanisms of exceptional human health span and life span. _Cold Spring Harb. Perspect. Med._ 13, a041204 (2023). Article CAS PubMed Google Scholar * Leung, Y. et
al. Cognition, function, and prevalent dementia in centenarians and near-centenarians: An individual participant data (IPD) meta-analysis of 18 studies. _Alzheimers Dement_ 19, 2265–2275
(2023). Article PubMed Google Scholar * Kenyon, C. J. The genetics of ageing. _Nature_ 464, 504–512 (2010). Article ADS CAS PubMed Google Scholar * Suh, Y. et al. Functionally
significant insulin-like growth factor I receptor mutations in centenarians. _Proc. Natl Acad. Sci._ 105, 3438–3442 (2008). Article ADS CAS PubMed PubMed Central Google Scholar *
Deelen, J. et al. A meta-analysis of genome-wide association studies identifies multiple longevity genes. _Nat. Commun._ 10, 3669 (2019). Article ADS PubMed PubMed Central Google Scholar
* Timmers, P. R. et al. Genomics of 1 million parent lifespans implicates novel pathways and common diseases and distinguishes survival chances. _eLife_ 8, e39856 (2019). Article PubMed
PubMed Central Google Scholar * Kaplanis, J. et al. Quantitative analysis of population-scale family trees with millions of relatives. _Science_ 360, 171–175 (2018). Article ADS CAS
PubMed PubMed Central Google Scholar * Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. _Nature_ 536, 285–291 (2016). Article CAS PubMed PubMed Central
Google Scholar * Shindyapina, A. V. et al. Germline burden of rare damaging variants negatively affects human healthspan and lifespan. _eLife_ 9, e53449 (2020). Article PubMed PubMed
Central Google Scholar * Ference, B. A. et al. Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. _N. Engl. J. Med._ 375, 2144–2153 (2016). Article CAS PubMed
Google Scholar * Jørgensen, A. B., Frikke-Schmidt, R., Nordestgaard, B. G. & Tybjærg-Hansen, A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. _N. Engl. J.
Med._ 371, 32–41 (2014). Article PubMed Google Scholar * Gutman, D. et al. Similar burden of pathogenic coding variants in exceptionally long‐lived individuals and individuals without
exceptional longevity. _Aging Cell_ 19, e13216 (2020). Article CAS PubMed PubMed Central Google Scholar * Lin, J.-R. et al. Rare genetic coding variants associated with human longevity
and protection against age-related diseases. _Nat. Aging_ 1, 783–794 (2021). Article PubMed Google Scholar * Karczewski, K. J. et al. Systematic single-variant and gene-based association
testing of thousands of phenotypes in 394,841 UK Biobank exomes. _Cell Genomics_ 2, 100168 (2022). Article CAS PubMed PubMed Central Google Scholar * Võsa, U. et al. Large-scale cis-
and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. _Nat. Genet._ 53, 1300–1310 (2021). Article PubMed PubMed Central
Google Scholar * Peters, M. J. et al. The transcriptional landscape of age in human peripheral blood. _Nat. Commun._ 6, 8570 (2015). Article CAS PubMed Google Scholar * Tyshkovskiy, A.
et al. Transcriptomic Hallmarks of Mortality Reveal Universal and Specific Mechanisms of Aging, Chronic Disease, and Rejuvenation. 2024.07.04.601982 Preprint at
https://doi.org/10.1101/2024.07.04.601982 (2024). * Tyshkovskiy, A. et al. Identification and application of gene expression signatures associated with lifespan extension. _Cell Metab._ 30,
573–593.e8 (2019). Article CAS PubMed PubMed Central Google Scholar * Liu, J. Z. et al. The burden of rare protein-truncating genetic variants on human lifespan. _Nat. Aging_ 2, 289–294
(2022). Article ADS CAS PubMed PubMed Central Google Scholar * Lagunas-Rangel, F. A. G protein-coupled receptors that influence lifespan of human and animal models. _Biogerontology_
23, 1–19 (2022). Article PubMed Google Scholar * Zhang, Z. et al. Increased hyaluronan by naked mole-rat Has2 improves healthspan in mice. _Nature_ 621, 196–205 (2023). Article ADS CAS
PubMed PubMed Central Google Scholar * Houtkooper, R. H. et al. Mitonuclear protein imbalance as a conserved longevity mechanism. _Nature_ 497, 451–457 (2013). Article ADS CAS PubMed
PubMed Central Google Scholar * Sebastiani, P. et al. Genetic signatures of exceptional longevity in humans. _PloS One_ 7, e29848 (2012). Article ADS CAS PubMed PubMed Central
Google Scholar * Jun, I. et al. ANO9/TMEM16J promotes tumourigenesis via EGFR and is a novel therapeutic target for pancreatic cancer. _Br. J. Cancer_ 117, 1798–1809 (2017). Article CAS
PubMed PubMed Central Google Scholar * Moqri, M. et al. Biomarkers of aging for the identification and evaluation of longevity interventions. _Cell_ 186, 3758–3775 (2023). Article CAS
PubMed PubMed Central Google Scholar * Moqri, M. et al. Validation of biomarkers of aging. _Nat. Med_. 1–13 (2024) https://doi.org/10.1038/s41591-023-02784-9. * Daunay, A. et al.
Centenarians consistently present a younger epigenetic age than their chronological age with four epigenetic clocks based on a small number of CpG sites. _Aging_ 14, 7718–7733 (2022).
Article PubMed PubMed Central Google Scholar * Ying, K. et al. A Unified Framework for Systematic Curation and Evaluation of Aging Biomarkers. 2023.12.02.569722 Preprint at
https://doi.org/10.1101/2023.12.02.569722 (2024). * Ying, K. et al. _ClockBase_: a comprehensive platform for biological age profiling in human and mouse. Preprint at
https://doi.org/10.1101/2023.02.28.530532 (2023). * Lu, A. T. et al. DNA methylation GrimAge version 2. _Aging_ 14, 9484–9549 (2022). CAS PubMed PubMed Central Google Scholar * Belsky,
D. W. et al. DunedinPACE, a DNA methylation biomarker of the pace of aging. _eLife_ 11, e73420 (2022). Article CAS PubMed PubMed Central Google Scholar * Ying, K. et al.
Causality-enriched epigenetic age uncouples damage and adaptation. _Nat. Aging_ 1–16 (2024) https://doi.org/10.1038/s43587-023-00557-0. * Timmers, P. R. H. J., Wilson, J. F., Joshi, P. K.
& Deelen, J. Multivariate genomic scan implicates novel loci and haem metabolism in human ageing. _Nat. Commun._ 11, 3570 (2020). Article ADS CAS PubMed PubMed Central Google
Scholar * Freudenberg-Hua, Y. et al. Disease variants in genomes of 44 centenarians. _Mol. Genet. Genom. Med._ 2, 438–450 (2014). Article CAS Google Scholar * Ryu, S. et al. Genetic
signature of human longevity in PKC and NF-κB signaling. _Aging Cell_ 20, e13362 (2021). Article CAS PubMed PubMed Central Google Scholar * Simon, M. et al. A rare human centenarian
variant of SIRT6 enhances genome stability and interaction with Lamin A. _EMBO J._ 42, e113326 (2023). Article CAS PubMed PubMed Central Google Scholar * Trajanovski, S.,
Martín-Hernández, J., Winterbach, W. & Van Mieghem, P. Robustness envelopes of networks. _J. Complex Netw._ 1, 44–62 (2013). Article Google Scholar * Bamshad, M. J. et al. Exome
sequencing as a tool for Mendelian disease gene discovery. _Nat. Rev. Genet._ 12, 745–755 (2011). Article CAS PubMed Google Scholar * Li, H. & Durbin, R. Fast and accurate short read
alignment with Burrows-Wheeler transform. _Bioinforma. Oxf. Engl._ 25, 1754–1760 (2009). Article CAS Google Scholar * McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce
framework for analyzing next-generation DNA sequencing data. _Genome Res_ 20, 1297–1303 (2010). Article CAS PubMed PubMed Central Google Scholar * Anderson, C. A. et al. Data quality
control in genetic case-control association studies. _Nat. Protoc._ 5, 1564–1573 (2010). Article CAS PubMed PubMed Central Google Scholar * Ng, P. C. & Henikoff, S. Predicting
deleterious amino acid substitutions. _Genome Res_ 11, 863–874 (2001). Article CAS PubMed PubMed Central Google Scholar * Adzhubei, I. A. et al. A method and server for predicting
damaging missense mutations. _Nat. Methods_ 7, 248–249 (2010). Article CAS PubMed PubMed Central Google Scholar * Chun, S. & Fay, J. C. Identification of deleterious mutations
within three human genomes. _Genome Res_ 19, 1553–1561 (2009). Article CAS PubMed PubMed Central Google Scholar * Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D.
MutationTaster2: mutation prediction for the deep-sequencing age. _Nat. Methods_ 11, 361–362 (2014). Article CAS PubMed Google Scholar * Lee, S., Abecasis, G. R., Boehnke, M. & Lin,
X. Rare-variant association analysis: study designs and statistical tests. _Am. J. Hum. Genet._ 95, 5–23 (2014). Article CAS PubMed PubMed Central Google Scholar * Zenin, A. et al.
Identification of 12 genetic loci associated with human healthspan. _Commun. Biol._ 2, 41 (2019). Article PubMed PubMed Central Google Scholar * Atkins, J. L. et al. A genome‐wide
association study of the frailty index highlights brain pathways in ageing. _Aging Cell_ 20, e13459 (2021). Article CAS PubMed PubMed Central Google Scholar * Timmers, P. R. H. J. et
al. Mendelian randomization of genetically independent aging phenotypes identifies LPA and VCAM1 as biological targets for human aging. _Nat. Aging_ 2, 19–30 (2022). Article PubMed Google
Scholar * Burgess, S., Zuber, V., Valdes‐Marquez, E., Sun, B. B. & Hopewell, J. C. Mendelian randomization with fine‐mapped genetic data: Choosing from large numbers of correlated
instrumental variables. _Genet. Epidemiol._ 41, 714–725 (2017). Article PubMed PubMed Central Google Scholar * Moqri, M. et al. Integrative epigenetics and transcriptomics identify aging
genes in human blood. 2024.05.30.596713 Preprint at https://doi.org/10.1101/2024.05.30.596713 (2024). * Tyshkovskiy, A. et al. Distinct longevity mechanisms across and within species and
their association with aging. _Cell_ 186, 2929–2949.e20 (2023). Article CAS PubMed PubMed Central Google Scholar * Ying, K. Centenarian genetic burden code. Zenodo
https://doi.org/10.5281/zenodo.13756349 (2024). Download references ACKNOWLEDGEMENTS We thank members of the Gladyshev laboratory for the discussions. This study was supported by NIH R01
AG064223 to V.N.G., and R01AG061155 and P01AG017242 to N.B. K.Y. was supported by NIH F99AG088431. The content is solely the responsibility of the authors and does not necessarily represent
the official views of the National Institutes of Health. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Division of Genetics, Department of Medicine, Brigham and Women’s Hospital and Harvard
Medical School, Boston, USA Kejun Ying, José P. Castro, Anastasia V. Shindyapina, Alexander Tyshkovskiy, Mahdi Moqri, Ludger J. E. Goeminne & Vadim N. Gladyshev * T. H. Chan School of
Public Health, Harvard University, Boston, USA Kejun Ying * i3S, Instituto de Investigação e Inovação em Saúde, Universidade do Porto and Aging and Aneuploidy Laboratory, IBMC, Instituto de
Biologia Molecular e Celular, Universidade do Porto, Porto, Portugal José P. Castro * Retro Biosciences, Redwood City, USA Anastasia V. Shindyapina * Department of Genetics, Albert Einstein
College of Medicine, Bronx, USA Sofiya Milman, Zhengdong D. Zhang & Nir Barzilai * Department of Medicine, Albert Einstein College of Medicine, Bronx, USA Sofiya Milman & Nir
Barzilai Authors * Kejun Ying View author publications You can also search for this author inPubMed Google Scholar * José P. Castro View author publications You can also search for this
author inPubMed Google Scholar * Anastasia V. Shindyapina View author publications You can also search for this author inPubMed Google Scholar * Alexander Tyshkovskiy View author
publications You can also search for this author inPubMed Google Scholar * Mahdi Moqri View author publications You can also search for this author inPubMed Google Scholar * Ludger J. E.
Goeminne View author publications You can also search for this author inPubMed Google Scholar * Sofiya Milman View author publications You can also search for this author inPubMed Google
Scholar * Zhengdong D. Zhang View author publications You can also search for this author inPubMed Google Scholar * Nir Barzilai View author publications You can also search for this author
inPubMed Google Scholar * Vadim N. Gladyshev View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS V.N.G. and K.Y. conceived the project. K.Y.
conducted the main data analysis. J.P.C., A.V.S., A.T., M.M., and L.J.E.G. assisted with data analysis. S.M. and Z.D.Z. provided clinical samples and data. N.B. supervised the clinical
aspects of the study. V.N.G. supervised the project. K.Y. and V.N.G. drafted the manuscript with input from all authors. All authors reviewed and approved the final version of the
manuscript. CORRESPONDING AUTHOR Correspondence to Vadim N. Gladyshev. ETHICS DECLARATIONS COMPETING INTERESTS After the initiation of this project, A.V.S. had a change in employment status
(Retro Biosciences). Analysis work was completed before this employment change. The other authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_
thanks Harold Bae and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE
DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed
under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or
format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material.
You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the
article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use
is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ying, K., Castro, J.P., Shindyapina, A.V. _et al._ Depletion of
loss-of-function germline mutations in centenarians reveals longevity genes. _Nat Commun_ 15, 9030 (2024). https://doi.org/10.1038/s41467-024-52967-2 Download citation * Received: 05 April
2024 * Accepted: 27 September 2024 * Published: 19 October 2024 * DOI: https://doi.org/10.1038/s41467-024-52967-2 SHARE THIS ARTICLE Anyone you share the following link with will be able to
read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing
initiative