A hybrid electro-thermochemical device for methane production from the air
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Coupling direct air capture (DAC) with methane (CH4) production is a potential strategy for fuel production from the air. Here, we report a hybrid electro-thermochemical device for
direct CH4 production from air. The proposed device features the cogeneration of carbon dioxide (CO2) and hydrogen (H2) in a single compartment via a bipolar membrane electrodialysis module,
avoiding a separate water electrolyzer, followed by a thermochemical methanation reaction to produce CH4. H2-induced disturbances lead to efficient CO2 extraction without pumping
requirement. The energy consumption and techno-economic analysis predict an energy reduction of 37.8% for DAC and a cost reduction of 36.6% compared with the decoupled route, respectively.
Accordingly, CH4 cost is reduced by 12.6%. Our proof-of-concept experiments show that the energy consumption for CO2 release and H2 production is 704.0 kJ mol−1 and 967.4 kJ mol−1,
respectively with subsequent methanation achieving a 97.3% conversion of CO2 and a CH4 production energy of 5206.4 kJ mol−1 showing a promising pathway for fuel processing from the air.
SIMILAR CONTENT BEING VIEWED BY OTHERS ENERGY COMPARISON OF SEQUENTIAL AND INTEGRATED CO2 CAPTURE AND ELECTROCHEMICAL CONVERSION Article Open access 14 September 2022 TOWARD ECONOMICAL
APPLICATION OF CARBON CAPTURE AND UTILIZATION TECHNOLOGY WITH NEAR-ZERO CARBON EMISSION Article Open access 05 December 2022 GAS DIFFUSION ELECTRODES, REACTOR DESIGNS AND KEY METRICS OF
LOW-TEMPERATURE CO2 ELECTROLYSERS Article 17 February 2022 INTRODUCTION Accumulating carbon dioxide (CO2) emissions from burning fossil fuels has been recognized as the primary driver of
global warming. The atmospheric CO2 concentration has increased from a pre-industrial value of 280 ppm to 423 ppm in 2024 (according to the global monthly mean CO2 data in May 2024)1. To
return global temperatures to the optimal levels of the pre-industrial Holocene period, atmospheric CO2 concentrations must fall below 350 ppm2,3. Achieving this goal is only partially
possible by expanding the share of renewable energy in the market, additional efforts are needed to actively remove at least 550 GtCO2 (550 billion tons of CO2) from the atmosphere by the
end of this century2,3. Direct air capture (DAC) offers the benefits of capturing CO2 directly from the air, exhibiting high flexibility in deployment location4,5,6. Owing to the low
concentration of CO2 in the air, the energy consumption for DAC is high. For example, the intrinsic thermodynamic penalty for capturing CO2 directly from the air (400 ppm, 0.44 GJ per tCO2)
increases by a factor of 3.7 compared to a more concentrated source (flue gas at 12%, 0.12 GJ per tCO2) when producing a 1 bar stream of CO25,7. In addition, any real process will require
even more energy than the thermodynamic minimum. For example, the estimated energy consumption for CO2 capture from concentrated sources requires ~0.8–5.6 GJ per tCO28,9,10, and ~4.5–12.2 GJ
per tCO2 for DAC8,11,12,13,14. The electrochemical carbon capture methods for DAC show great promise in terms of both energy efficiency and cost effective, as the process occurs at ambient
reaction conditions and can harness the potential of integration with renewable electricity5,15,16. The captured CO2 can be either stored by geological storage17 or further converted to
value-added products18,19,20,21,22. The conversion of captured CO2 to renewable methane (CH4), the major component in natural gas, is of particular interest because (i) CH4 is an important
chemical raw material for various chemical products (e.g., olefins and aromatics)23, (ii) CH4 can be acted as a good hydrogen (H2) carrier due to matured storage and distribution network for
natural gas and (iii) CH4 can be directly used as fuel for heating and power generation. Hence, capturing and converting CO2 into renewable CH4 could facilitate the efficient and extensive
distribution of energy services with net carbon emission. Generally, renewable CH4 from sunlight, water, and CO2 can be generated by thermochemical and (photo)electrochemical methods.
Thermochemical method using an energy downhill CO2 methanation reaction, also known as the Sabatier reaction CO2 + 4H2 → CH4 + 2H2O, is thermodynamically favorable, which yields high
conversion and selectivity21,24 and requires green H2 as the feedstock. Due to its energy-downhill nature, the reaction can operate in a self-sustainable manner without additional energy
input for continuous reaction. Furthermore, the Sabatier reaction shows advantageous thermodynamic properties, reaching a carbon conversion efficiency > 90% under a typical operating
conditions of 1 bar and 300 °C24. While the (photo)electrochemical method requires the captured CO2 to undergo a deep reduction process, i.e., 8 electrons reduction, leading to unfavorable
reaction kinetics and low selectivity21,25,26. One recent work be Cao-Thang Dinh group reported a bicarbonate-fed electrochemical system using a bipolar membrane (BPM) that achieved a
CO2-to-CH4 conversion with over 70% Faradaic efficiency at a current density range of 100–750 mA cm−2 by alternating current operation27. Although 70% Faradic efficiency for CH4 is rather
high by electrochemical reactions, additional exhaust treatment is needed to recover the major by-product of H2 and unreacted CO2. Hence, the benefits of electrochemical DAC and
thermochemical CO2 methanation can be combined to achieve a highly efficient as well as cost-effective DAC and its subsequent conversion into renewable CH4. In this study, we present a
proof-of-concept hybrid electro-thermochemical device that integrates an electrochemical module for simultaneous DAC and H2 production cascaded with a thermochemical reactor to produce CH4.
The electrochemical module based on bipolar membrane electrodialysis (BPMED) operates at room temperature and only uses electricity as the energy input. As H2 and CO2 are released within the
same electrode compartment (i.e., cathode compartment), the co-generated H2 can serve as a sweep gas for CO2 by reducing CO2 partial pressure and facilitating its release. This strategy
further avoids the use of vacuum pumping for CO2 extraction, leading to reduced energy consumption and system cost. Particularly, the electrochemical BPMED module enables simultaneous CO2
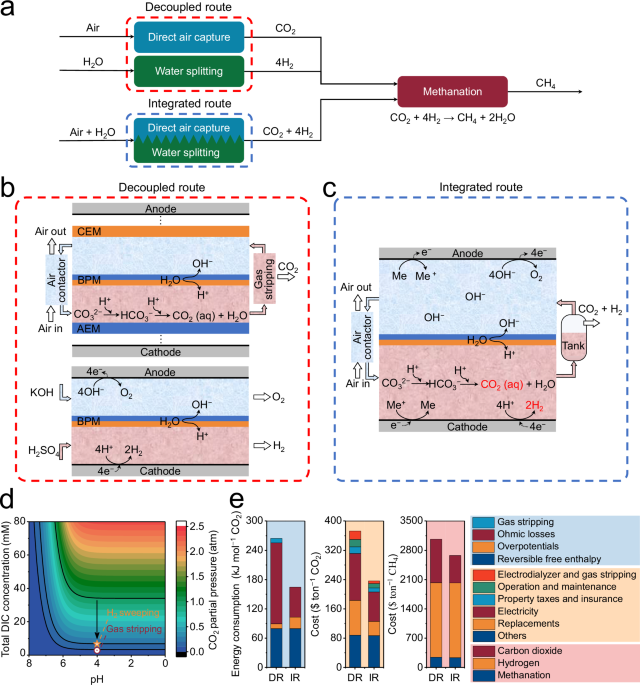
release and H2 production with tunable ratios (e.g., 1: 4 CO2 to H2) intended for subsequent methanation reaction. RESULTS INTEGRATED ROUTE CONCEPT As shown in Fig. 1a, the CH4 production
consists of two cascaded steps in general: (i) DAC and water splitting for CO2 release and H2 production and (ii) CO2 methanation via a methanation reactor. The first step can be achieved
either by a decoupled route in which DAC and water splitting are performed in separate reactors (red box in Fig. 1a) or an integrated route in which DAC and water splitting are performed in
a single integrated reactor (blue box in Fig. 1a). Figure 1b,c show the schematic illustration of decoupled and integrated routes. The decoupled route consists of a separate DAC module and a
separate water-splitting module (red box in Fig. 1b). The separate DAC module is based on the pH-swing method via a BPMED cell. When a sufficient electric field is applied, the BPM
dissociates water into hydroxide (OH−) and proton (H+) ion, producing a controllable _∆_pH over the two sides of the membrane28. The OH− ions and H+ ions produced by BPMED can be used for
CO2 absorption and CO2 release, respectively. The compartments were separated a cation exchange membrane (CEM) or anion exchange membrane (AEM). Note that, to enhance CO2 release, extracting
of CO2 from the acidifying compartment requires additional gas stripping (red box in Fig. 1b) using vacuum-assisted stripping devices (including a vacuum pump, membrane contactor, and so
on). A separate BPMED is shown as an example in Fig. 1b (red box) to represent typical water electrolysis. Note that water electrolysis is a well-developed technology for the conversion of
water into H2 and oxygen (O2) and can be realized by various membrane technologies, e.g., BPM29,30,31,32,33, AEM34,35, and proton exchange membrane (PEM)34,35. The voltage requirements of
BPM, AEM, and PEM electrolyzers are similar36. In this study, we only take BPM-based electrolysis technology as an example for direct comparison with our proposed integrated route concept.
In addition, using a BPM could break the electrocatalysts’ pH incompatibility, allowing for the separate optimization of catalyst/electrolyte pairing of the two half-reactions of water
splitting irrespective of the counter electrodes environment, enabling performance enhancement potentials32. In this study, we proposed an integrated route concept, shown in Fig. 1c, which
utilizes a single BPM-based electrochemical reactor to perform DAC and water splitting simultaneously. An aqueous fast redox couple, e.g., potassium ferro/ferricyanide (K3/K4[Fe(CN)6]), is
utilized to replace the water-splitting reaction, i.e., hydrogen evolution reaction (HER) at the cathode and oxygen evolution reaction (OER) at the anode37. Due to a more positive
equilibrium potential of 0.358 V (vs. SHE) than HER of −0.561 V (vs. SHE) and a more negative equilibrium potential of 0.358 V (vs. SHE) than OER of 0.669 V (vs. SHE) at pH = 9.5, in combine
with fast intrinsic kinetics, the reversible K3/K4[Fe(CN)6] reaction is preferred at two electrodes of the integrated route. The water-splitting reaction happens only when the
K3/K4[Fe(CN)6] redox couples depleted at high current densities. Ideally, at high current densities, the H+ ions generated through BPM are partly utilized for CO2 release and the other part
for the H2 production. Hence, the ratio of CO2 to H2 can be precisely controlled by regulating the current density and the concentration of K3/K4[Fe(CN)6], under the condition of a fixed
dissolved inorganic carbon (DIC) input rate. In addition, since CO2 and H2 are simultaneously generated on the cathode compartment (Fig. 1c), H2 can be utilized as a sweep gas to decrease
the partial pressure of CO2 as well as additional disturbance to the electrolyte due to H2 bubble movement, avoiding the application of vacuum stripping, as shown in Fig. 1d. Based on the
required ratio of CO2 and H2 of 1: 4 in the methanation reactor for methanation, the partial pressure of CO2 can be reduced from 1.0 to 0.2 atm, and its effect is similar to that of vacuum
stripping, as shown in Fig. 1d. Compared to decoupled route, the energy consumption of integrated route for CO2 release is reduced by 37.8% (Fig. 1e) due to the decrease in ohmic losses of
membranes and the avoidance of vacuum-assisted gas stripping devices (Supplementary Note 1 for calculations). Meanwhile, the simplified integrated route (Fig. 1c), compared to the decoupled
route (Fig. 1b), leads to cost reductions from 372.7 $ ton−1 CO2 to 236.3 $ ton−1 CO2, a 36.6% reduction (Fig. 1e). The techno-economic analysis also revealed that a 12.6% reduction (from
3065.0 $ ton−1 CH4 to 2679.3 $ ton−1 CH4) in the cost of CH4 production via integrated route is attributed to decreased carbon capture cost (Fig. 1e). The detailed assumptions for the
techno-economic analysis of technologies evaluated in this study are included in the Supplementary Note 2 and Supplementary Data 1–4, and the database files used for arriving at detailed
cost values are also included in the Supplementary Data 1–4. In summary, the comparison between the decoupled route and the integrated route reveals that the structure of the integrated
route possesses the following advantages: (i) Energy consumption reduction: Cell structure is simplified as fewer membranes and compartments are used. Therefore, the low resistivity of the
device leads to a low overall process energy consumption. (ii) Cost reduction: The simplified cell structure and the avoidance of vacuum-assisted gas stripping devices also contribute to
cost reduction. (iii) Easier operational control: the integrated route enables simultaneous CO2 release and H2 production with tunable ratios (e.g., 1: 4 CO2 to H2) intended for subsequent
methanation reaction. Furthermore, the feasibility of the integrated route has been validated via our dedicated experiments (Supplementary Note 3). An experimental demonstration of the
proposed integrated route for proof-of-concept purposes is shown in the following section. HYBRID ELECTRO-THERMOCHEMICAL DEVICE Figure 2a shows the schematic illustration of the BPMED module
for CO2 release and H2 production. The BPMED module contained two compartments (i.e., anode compartment and cathode compartment) separated by a BPM, and two electrodes for electrochemical
reactions, i.e., fast redox reactions of K3/K4[Fe(CN)6] and water splitting reactions. At the middle of the BPMED module, a BPM that generates H+ and OH− ion fluxes via water dissociation
reaction (Eq. (1)) at the BPM interface was used to convert the input electrolytes into acidified steam for CO2 release (Eq. (2)) and basified stream for CO2 absorption (Eq. (3)),
respectively. The electrode reactions at low current densities were majorly one electron, reversible redox reactions (Eq. (4) and Eq. (5)). The increase of current density led to the
depletion of the fast redox couple, subsequently triggering the oxygen evolution reaction (OER) at the anode (Eq. (6)) and hydrogen evolution reaction (HER) at the cathode (Eq. (7)).
Ideally, at high current densities, the H+ ions generated through water dissociation are partly utilized for CO2 release and the other part for the H2 production. Hence, the ratio of CO2 to
H2 can be precisely controlled by regulating the current density and the concentration of K3/K4[Fe(CN)6], under the condition of a fixed DIC rate. $${{\rm{Water\;
dissociation}}}\,({{\rm{BPM}}}):{{{\rm{H}}}}_{2}{{\rm{O}}}\to {{{\rm{H}}}}^{+}+{{{\rm{OH}}}}^{-}$$ (1) $${{{\rm{CO}}}}_{2}\,{{\rm{release}}}\,({{\rm{cathode\;
compartment}}}):{{{\rm{CO}}}}_{3}^{{2}^{-}}+{2{{\rm{H}}}}^{+}\leftrightarrow {{{\rm{HCO}}}}_{3}^{-}+{{{\rm{H}}}}^{+}\leftrightarrow {{{\rm{CO}}}}_{2}+{{{\rm{H}}}}_{2}{{\rm{O}}}$$ (2)
$${{{\rm{CO}}}}_{2}\,{{\rm{absorption}}}\,({{\rm{base\; tank}}}):{{{\rm{CO}}}}_{2}+{2{{\rm{OH}}}}^{-}\leftrightarrow {{{\rm{HCO}}}}_{3}^{-}+{{{\rm{OH}}}}^{-}\leftrightarrow
{{{\rm{CO}}}}_{3}^{{2}^{-}}+{{{\rm{H}}}}_{2}{{\rm{O}}}$$ (3) $${{\rm{Anode}}}:{\left[{{\rm{Fe}}}{\left({{\rm{CN}}}\right)}_{6}\right]}^{{4}^{-}}\to
{\left[{{\rm{Fe}}}{\left({{\rm{CN}}}\right)}_{6}\right]}^{{3}^{-}}+{{{\rm{e}}}}^{-}$$ (4)
$${{\rm{Cathode}}}:{\left[{{\rm{Fe}}}{\left({{\rm{CN}}}\right)}_{6}\right]}^{{3}^{-}}+{{{\rm{e}}}}^{-}\to {\left[{{\rm{Fe}}}{\left({{\rm{CN}}}\right)}_{6}\right]}^{{4}^{-}}$$ (5)
$${{\rm{Anode}}} \, ({{\rm{OER}}} \, {{\rm{in}}} \, {{\rm{alkaline}}} \, {{\rm{solution}}}):{4{{\rm{OH}}}}^{-}\to {{{\rm{O}}}}_{2}+{2{{\rm{H}}}}_{2}{{\rm{O}}}+{4e}^{-}$$ (6)
$${{\rm{Cathode}}} \, ({{\rm{HER}}} \, {{\rm{in}}} \, {{\rm{acidic}}} \, {{\rm{solution}}}):{2{{\rm{H}}}}^{+}+{2{{\rm{e}}}}^{-}\to {{{\rm{H}}}}_{2}$$ (7)
$${{{\rm{CO}}}}_{2}\,{{\rm{methanation}}}\,({{\rm{methanation\; reactor}}}):{{{\rm{CO}}}}_{2}+{4{{\rm{H}}}}_{2}\to {{{\rm{CH}}}}_{4}+{2{{\rm{H}}}}_{2}{{\rm{O}}}$$ (8) $${{\rm{Overall\;
reaction}}}:{{{\rm{CO}}}}_{2}+{2{{\rm{H}}}}_{2}{{\rm{O}}}\to {{{\rm{CH}}}}_{4}+{2{{\rm{O}}}}_{2}$$ (9) Figure 2b shows the schematics of the CH4 production using the hybrid
electro-thermochemical device, which consists of three modules: (i) CO2 absorption module (blue box), (ii) BPMED module (orange box), and (iii) thermochemical methanation module (red box).
The CO2 absorption module utilized an air pump to introduce air into the base tank for CO2 absorption from air. The CO2 was absorbed in the base tank. Subsequently, the base electrolyte was
introduced into the electrolyte tank to mix with the acid electrolyte from the acid tank, and then pumped to the BPMED module. In the BPMED module, the electrolyte was acidified in the
cathode compartment and basified in the anode compartment. The basified stream and O2 were directed toward the base tank to separate O2 and base electrolyte, and the base electrolyte will be
again used for CO2 absorption from the air. The acidified stream was directed to the acid tank to separate the gas mixture (i.e., CO2 and H2) and acid electrolyte. The acid electrolyte was
then introduced into the electrolyte tank. The gas mixture separated from the acid tank was directly introduced into the thermochemical methanation module to produce CH4. (Eq. (8)). The
overall reaction of the hybrid electro-thermochemical device is shown in Eq. (9). To simplify the operation, initially, the CO2 absorption process was replaced by a simulated equilibrium
electrolyte. The electrolytes comprised of 100-300 mM K3/K4[Fe(CN)6], 54.32 mM KHCO3, and 22.80 mM K2CO3 (i.e., 100 mM KOH in equilibrium with 400 ppm CO2, see Supplementary Note 4). Based
on these premises, the CH4 production performance of the hybrid electro-thermochemical device was further tested under real DAC conditions. In actual operation, two recirculation loops were
added for the anode and cathode compartments to increase the flow speed inside the BPMED module, respectively, as shown in Supplementary Note 5. The flow rates of the electrolyte and
recirculation were 2 ml min−1 and 30 ml min−1, respectively. The methanation reactor temperature was regulated through ceramic heating at 320 °C. PERFORMANCES OF BPMED MODULE FOR CO2 RELEASE
AND H2 PRODUCTION We first explored the effect of K3/K4[Fe(CN)6] concentration on CO2 release and H2 production in the BPMED module. Figure 3a, b show the carbon removal efficiency as a
function of current density during acidification. At a high K3/K4[Fe(CN)6] concentration of 300 mM and a low current density of 10 mA cm−2, the HER and OER at the electrodes were completely
replaced by the fast redox reactions of K3/K4[Fe(CN)6] due to its favorable thermodynamics and reaction kinetics, as shown in Fig. 3c (no H2 production). Hence, the H+ fluxes generated at
the BPM interface were mostly used to convert the input electrolyte into an output stream of acidified electrolyte (pH > 7.7 ± 1.1), i.e., converting CO32− to HCO3− and CO2 (aq),
resulting in a detectable release of CO2 (g) (Fig. 3c) and a low carbon removal efficiency of 4.7 ± 3.4% (Fig. 3a). Therefore, at low current densities ( < 10 mA cm−2), the release of CO2
can be attributed to the pH-induced equilibrium shift. As shown in Fig. 3a, the carbon removal efficiency gradually increased from 4.7 ± 3.4% to 80.9 ± 17.6% when the current density was
increased from 10 to 40 mA cm−2, at a high K3/K4[Fe(CN)6] concentration of 300 mM. Note that, in the current density range of 10 to 40 mA cm−2, the pH of the acidified stream decreased from
7.7 ± 1.1 to 4.0 ± 1.2 (Fig. 3b), and the competing water splitting reaction started to occur due to the consumption of K3/K4[Fe(CN)6] redox-couple (Fig. 3c). The decreased pH and the
produced H2 result in an increase of CO2 (aq) concentration and a decrease of CO2 partial pressure in the cathode compartment respectively. These changes promote the release of CO2 (g) from
an acid electrolyte. At a high K3/K4[Fe(CN)6] concentration of 300 mM, and high current densities >40 mA cm−2, the pH of the acidified stream was <4.0 ± 1.2 (Fig. 3b), indicating that
most of the carbon species in the acid electrolyte exist as CO2 (aq) (Supplementary Fig. 5b). As a result, at this stage (current densities >40 mA cm−2), the increase in carbon removal
efficiency (Fig. 3a) was mainly due to the significant release of H2 (Fig. 3c, d), resulting in a further reduction in the partial pressure of CO2. The partial pressure of CO2 reduced from
0.59 to 0.20 atm when increasing the current density from 40 to 120 mA cm−2 (Fig. 3c). Hence, we observed a further increase in carbon removal efficiency from 80.9 ± 17.6% to 96.0 ± 9.5% at
a current density of 120 mA cm−2 (Fig. 3a), which we attributed to the H2-induced CO2 release as a result of reduced partial pressure of CO2. We observed that the concentration of reversible
redox couple (K3/K4[Fe(CN)6]) in the electrolyte significantly affects the carbon removal efficiency, as shown in Fig. 3a. In general, higher K3/K4[Fe(CN)6] concentration leads to higher
carbon removal efficiency due to fast accumulation of H+ content in the cathode compartment, as shown in Fig. 3b. At a low K3/K4[Fe(CN)6] concentration (e.g., 100 mM), the carbon removal
efficiency was only 20.0 ± 4.1% at 40 mA cm−2 which was a result of high pH values (Fig. 3b) resulting from earlier dominating of water splitting reactions (Fig. 3c). In the absence of
K3/K4[Fe(CN)6], water splitting reaction at the electrodes is the main reaction (see Supplementary Fig. 10). As shown in Fig. 3d, the released ratio of H2 to CO2 can be regulated by
manipulating the operation current density and the concentration of K3/K4[Fe(CN)6] at a fixed DIC rate input to the BPMED device. In general, higher current density leads to higher H2/CO2
ratios due to a gradually increased water-splitting reaction. Note that at high concentrations of K3/K4[Fe(CN)6], the increase in the slope of the curves at high current density can be
explained by the CO2 release reaching its maximum (due to depletion of K3/K4[Fe(CN)6]) while HER is the major reaction with further increase in current density. As shown in Fig. 3d, at
concentrations of 100 mM, 200 mM, and 300 mM K3/K4[Fe(CN)6], the current density can be precisely regulated to achieve values of 0–20 mA cm−2, 100–120 mA cm−2 and 120 mA cm−2
correspondingly, thereby enabling adjustment of the H2 to CO2 ratio to a desired level of 4: 1 which can be further utilized for methanation reaction. The corresponding current efficiency is
shown in Fig. 3e. We observed that the current efficiency is not 100% by only considering CO2 release and H2 production. At low current densities, the lost current efficiency can be
attributed to (i) the leak current due to the imperfect selectivity of the cation exchange layer (CEL) and anion exchange layer (AEL) of the BPM37, (ii) H+ (changing pH), and (iii) H+
participating in the formation of HCO3− and H2CO3, as shown in Supplementary Note 6. At high current densities, the partial current density in BPM due to co-ion leakage is minor, and most of
H+ produced by BPM is utilized for the release of CO2 and H2, hence leading to a close to 100% current efficiency as shown in Fig. 3e. Figure 3f shows the total cell voltage (solid lines)
and energy consumption (bars) as a function of the current density using different K3/K4[Fe(CN)6] concentrations. The cell voltages increased with the current density and decreased with the
rising K3/K4[Fe(CN)6] concentration. We measured the energy consumption for CO2 release increased from 372.3 ± 18.4 to 551.0 ± 7.6 kJ mol−1 at 100 mM K3/K4[Fe(CN)6] with the increase of
current density from 10 to 40 mA cm−2, and 119.4 ± 1.4 kJ mol−1 to 640.1 ± 40.0 kJ mol−1 at 300 mM M K3/K4[Fe(CN)6] with the increase of the current density from 10 to 140 mA cm−2. Note that
there is a maximum CO2 output rate with increasing current density due to the depletion of DIC. Further increase in current density led to no additional CO2 release (Fig. 3c). For example,
the CO2 output rate reached 3.4 ± 0.4 ml min−1 at 80 mA cm−2 and minor changes, i.e., 3.5 ± 0.2 ml min−1, at 140 mA cm−2 with 300 mM K3/K4[Fe(CN)6]. While the H2 output rate was continuously
increased with the increasing current density. The measured H2 output rate increased from 0.2 ± 0.1 to 17.2 ± 0.4 ml min−1 at 300 mM K3/K4[Fe(CN)6] when increasing the current density from
20 to 140 mA cm−2. CASCADED THERMOCHEMICAL METHANATION We demonstrated a proof-of-concept device for cogeneration of CO2 and H2 with subsequent direct conversion into CH4. The simulated
electrolyte consisting of 54.32 mM KHCO3, 22.80 mM K2CO3 and 300 mM K3/K4[Fe(CN)6] as the electrolyte input to the BPMED module at a current density of 140 mA cm−2. As shown in Fig. 4a, the
rates of CO2 and H2 output stabilized at 3.7 ml min−1 and 17.0 ml min−1 after 40 min of operation, forming a 4.6: 1 ratio of H2 to CO2 (close to 4 required for methanation reaction, Fig.
4b). The presence of a slight excess of H2 can enhance the CO2 conversion efficiency and the CH4 selectivity38. After 60 min of operation, the gas mixture (i.e., CO2 and H2) produced by the
BPMED module was directed into the methanation module operating at 320 °C. The CH4 output rate stabilized at 3.4 ml min−1 after 20 min of methanation reaction. The CO2 conversion efficiency,
based on the measured gas composition (Fig. 4b), was found to be 91.1% (Fig. 4c). The electric energy consumption for the BPMED module to produce CH4 was 6412.3 kJ mol−1 CH4. Note that the
exhaust gas steam contains 48.7% of unreacted H2, i.e., 3.5 ml min−1 of exhaust H2 (equivalent to a H2 conversion of 79.5%, Fig. 4b). Further gas separation or enhanced reaction extent can
increase the energy efficiency of the methanation. The CH4 can be separated from the gas mixture by a membrane separator at a high purity level ( >99.9%)39. The permeate stream containing
H2, unreacted CO2 and a small amount of CH4 can be recycled back to the methanation module. The stability of the hybrid electro-thermochemical device was evaluated by subjecting it to
continuous operation for 30 h. After 6 h of operation, the gas mixture (i.e., CO2 and H2) produced by the BPMED module was directed into the methanation module operating at 320 °C. The
methanation reaction lasted for a duration of 22 h, after which the gas-producing composition of the BPMED module was remeasured. As shown in Fig. 4d, the output rate of CH4 was stably
produced from the methanation module during the operation time of the methanation reaction (i.e., from the 6th to the 28th hour), indicating that the output of CO2 and H2 from the BPMED
module was also stable. In addition, the output rates of CO2 and H2 from the BPMED module were re-measured during the operation time from the 28th to the 30th hour. As shown in Fig. 4d, the
output rates of CO2 and H2 remained stable during this period. Meanwhile, the cell voltage of the BPMED module remained consistently below 6.0 V for 30 h, demonstrating a sustained and
stable trend. The temperature of the methanation module remained stable (without any external intervention) during the operation of the methanation reaction (Fig. 4d). The potential for
practical CH4 production from the air was further demonstrated by the high stability of the hybrid electro-thermochemical device. THERMOCHEMICAL METHANATION UNDER REAL DAC CONDITIONS The CH4
production performance of the hybrid electro-thermochemical device was further tested under real DAC conditions. The real DAC process consists of two phases, namely CO2 absorption and CO2
release. The CO2 absorption was performed indoor by pumping air to the base tank. The base tank was filled with a 100 mM KOH solution, and the volume was 1000 ml. The flow rate of the air
pump was 1750 ± 90 ml·min−1 and was quantified by a mass flow meter. The air undergoes a dehydration process before entering the mass flow meter through condensation in order to prevent
potential damage to the mass flow meter during long periods of operation. The air from the mass flow meter was introduced into a bubbler immersed in the KOH solution. After approximately 83
h of CO2 absorption, a carbon-containing electrolyte of 860 ml was obtained, indicating a water volumetric loss of 14% compared to the initial 1000 ml KOH solution due to evaporation. As a
result, the total alkalinity of the electrolyte increases from 100 mM to 116.3 mM. As shown in Fig. 5a, the CO2 concentration of the base tank inlet and outlet was monitored by two CO2
sensors, respectively, while the pH of the base electrolyte was concurrently measured using a pH meter. The CO2 absorption process could be divided into four stages: fast absorption (0–22 h,
electrolyte: pH > 12), transition (22–40 h, electrolyte: 12 > pH > 10), slow absorption (40–65 h, electrolyte: 10 > pH > 9.8), and saturation ( > 65 h, electrolyte: pH ~
9.8). Subsequently, K3/K4[Fe(CN)6] was added to the saturation electrolyte. The concentration of K3/K4[Fe(CN)6] in the mixed electrolyte was 300 mM. The CO2 release and H2 production were
performed in the BPMED module at a current density of 140 mA cm−2, while the flow rates of the electrolyte and recirculation were 2 ml min−1 and 30 ml min−1, respectively. Figure 5b
indicates that after 50 mins of operation, both the output CO2 and H2 reached to relative stable values, i.e., ~4.1 ml min−1 for CO2 and ~18.6 ml min−1 for H2, forming an H2/CO2 of 4.5
(close to 4 required for methanation reaction). After 60 mins of operation, the outlet gas mixture (i.e., CO2 and H2) from the BPMED module was directly fed into the methanation reactor
operating at 320 °C. The CH4 output rate reached a stable value of ~4.0 ml min−1 after 32 mins of CO2 methanation. The average CO2 conversion efficiency was 97.3% after 54 mins of CO2
methanation (Supplementary Fig. 12a). Meanwhile, the total cell voltage remained constant for approximately 170 min, with an average of 5.05 V (Supplementary Fig. 12b). The combination of
low pH (3.5, Supplementary Fig. 12c) of the acidified stream and low CO2 partial pressure (0.18 atm) ensures a high carbon removal efficiency (95.3%). The pH of the basified stream reaches
13.0 simultaneously (Supplementary Fig. 12c), hence the base electrolyte can continue to absorb CO2 from the air (Fig. 5a). The Sankey diagram in Fig. 5c illustrates the energy breakdown
during the conversion process from electrical power input to CH4 (Supplementary Note 8 for the detailed calculations). The electrical power input can be divided into two main parts, namely
CO2 release energy consumption (orange box) and H2 production energy consumption (blue box), which account for 13.9% and 86.1% of the total energy consumption, respectively. Figure 5c also
shows that the overpotentials cover a significant part of total energy consumption, corresponding to 66.3% (12.2% for CO2 release and 54.1% for H2 production) of the electrical power input.
The higher overpotential may be attributed to the abundant generation of bubbles in the electrolyte and the poor performance of the electrocatalyst. Bubbles formed at the electrode can
reduce the active surface area and hinder ion transport in the electrolyte, leading to energy losses40. The dimensions of both the anode and cathode compartments in the BPMED device are 4.5
cm × 4.5 cm × 0.25 cm, while the outlet diameter was only 0.25 cm. The total gas output rate in both the anode and cathode compartments is approximately 32 ml min−1, i.e., 4.1 ml min−1 of
CO2, 18.6 ml min−1 of H2, and 9.3 ml min−1 of O2 (half the H2 output rate). Hence, the narrow compartments and the small outlet may prevent the timely discharge of bubbles generated by the
electrode from the compartments, resulting in an increase in overpotentials. The cell voltage can be effectively reduced by increasing the electrolyte flow rate in compartments, as shown in
Supplementary Fig. 8. Because the diameter of bubble detachment on the electrode surface is inversely proportional to the flow velocity40. On the other hand, in this study, we used the
commercial electrodes, i.e., Pt-coated titanium mesh as the cathode and Ti-coated ruthenium (Ru)-iridium (Ir) as the anode. These electrodes performed well for separate water splitting and
K3/K4[Fe(CN)6] (Supplementary Fig. 14). While a significant reduction in the HER electrochemical performance was observed with an electrolyte of 50 mM H2SO4 and 300 mM K3/K4[Fe(CN)6]
(Supplementary Fig. 14). The on-site potential of HER in the mixture of 50 mM H2SO4 and 300 mM K3/K4[Fe(CN)6] was negatively shifted by approximately 0.5 V compared to that in 50 mM H2SO4.
Hence, engineering better-performing electrocatalysts (and/or electrode structure) is also essential for the further enhancement of the integrated reactor. The energy loss caused by the
overpotentials account for 80.0% of total energy consumption (see Fig. 5c). Hence, we simply considered a reduced device overpotential to access the potential of the system with the Sankey
diagram for the energy consumption shown in Fig. 5d (Supplementary Note 8 for the detailed calculations). Based on the literature data, the overpotential of typical NiFe/nickel foam
(NiFe/NF) for OER at 140 mA cm−2 is estimated to be ~280 mV in a 1 M KOH solution41. The typical overpotential of CoP nanoparticles encapsulated in ultrathin nitrogen-doped porous carbon
(CoP@NC) as for HER at 140 mA cm−2 is estimated to be ~140 mV in a 0.5 M H2SO4 solution42. Meanwhile, the water dissociation overpotential, i.e., the water dissociation reaction in the
junction layer of the BPM, is assumed to be ~100 mV36. Assuming our device can operate at these H2 production performances, a 51.9% reduction (from 704.0 kJ mol−1 to 338.4 kJ mol−1) of CO2
release energy consumption and a 52.8% reduction (from 5206.4 kJ mol−1 to 2459.4 kJ mol−1) of CH4 production energy consumption can be achieved. DISCUSSION In this work, we present a
proof-of-concept hybrid electro-thermochemical device for direct CH4 production from an electrochemical BPMED module based on BPM with simultaneous DAC and H2 production cascaded with a
thermochemical methanation reactor. The electrochemical BPMED module relies on the principle of “pH-swing”16 to efficiently absorb and release CO2. Meanwhile, the “pH-swing” of the working
fluid enables an optimal acid/alkaline amphoteric electrolytic environment for efficient water splitting for H2 production. As H2 and CO2 are released within the same compartment, the H2 can
serve as sweep gas for CO2 release by reducing the CO2 partial pressure, avoiding the use of vacuum pumping for CO2 stripping. Meanwhile, cell structure is simplified as fewer membranes and
compartments are used. Therefore, the low resistivity of the system leads to a low overall process energy consumption. Reduced membrane quantities also contribute to cost reduction. The
energy consumption and techno-economic analysis predicted an energy reduction of 37.8% for DAC and a cost reduction of 34.5% compared with the decoupled pathway. Accordingly, CH4 cost was
reduced by 11.4%. Particularly, the electrochemical BPMED module proposed in this study enables the simultaneous production of CO2 and H2 gas with tunable ratios at different operation
current densities and the concentration of K3/K4[Fe(CN)6] under the condition of a fixed DIC rate, which can be used for subsequent hydrocarbon synthesis. We demonstrated the subsequent
methanation reaction with a CO2: H2 molar ratio of 1: 4. To make the study closer to real DAC, we prepared simulated electrolytes (mixtures of 54.32 mM KHCO3 and 22.80 mM K2CO3, i.e., 100 mM
KOH in equilibrium with 400 ppm CO2) as the electrolytes input to the module. The electrolytes were also simultaneously added with 100–300 mM K3/K4[Fe(CN)6]. The electrode reactions at low
current densities were majorly one electron, reversible redox reaction. Hence, the H+ produced by water dissociation in BPM was used to CO2 release, i.e., converting
\({{{\rm{CO}}}}_{3}^{{2}^{-}}\) to \({{{\rm{HCO}}}}_{3}^{-}\) and CO2. The initiation of the HER occurs with the further increase of current density ( >10 mA cm−2). Upon reaching a CO2 to
H2 gas production ratio of 1: 4, the energy consumption for CO2 release and H2 production amounts to 614.6 ± 58.5 kJ mol−1 and 948.8 ± 90.3 kJ mol−1, respectively, at a current density of
120 mA cm−2, an electrolyte flow rate of 2 ml min−1 and a K3/K4[Fe(CN)6] concentration of 300 mM. The hybrid electro-thermochemical device was used for CH4 production. The device exhibited
consistent CH4 output (3.6 ml min−1) and achieved a CO2 conversion efficiency of 96.1% during the long stability test. The hybrid electro-thermochemical device was used for CH4 production
under real DAC conditions. The device shown that the energy consumption for CO2 release and H2 production was 704.0 kJ mol−1) and 967.4 kJ mol−1 with subsequent methanation achieved a 97.3%
conversion of CO2 and a CH4 production energy of 5206.4 kJ mol−1 showing a promising pathway for fuel processing from air. The optimization of overpotential in the BPMED module was
conducted. A 51.9% reduction (from 704.0 kJ mol−1 to 338.4 kJ mol−1) of CO2 release energy consumption and a 52.8% reduction (from 5206.4 kJ mol−1 to 2459.4 kJ mol−1) of CH4 production
energy consumption can be achieved. Further improvement in the device performance requires better-performing BPMED module construction and electrocatalysts (and/or electrode structure)
engineering to reduce overpotentials in the BPMED module. The proof-of-concept device provides a promising pathway for direct CH4 production using co-produced CO2 and H2 from the air.
METHODS CHEMICALS Potassium ferricyanide (K3[Fe(CN)6], AR, ≥99.5%), potassium ferrocyanide trihydrate (K4[Fe(CN)6]·3H2O, AR, 99.0%), potassium bicarbonate (KHCO3, AR, 99.5%), potassium
carbonate (K2CO3, AR, 99%), potassium hydroxide (KOH, AR, 85%), ruthenium on alumina (extent of labeling: 5 wt.% loading, powder, reduced, dry), and fiberglass wool for laboratory use
(Shanghai Boer Chemical Reagents CO., Ltd). ELECTRODES The electrodes were commercially procured. The anode for the BPMED module was made of titanium (Ti) meshes with a coating of ruthenium
(Ru)-iridium (Ir), and the thickness of the coating is 8 μm. The cathode for the BPMED module was made of titanium (Ti) meshes with a platinum (Pt) coating, and the thickness of the coating
is 1 μm. Product information was provided by the merchant. The dimensions of the electrodes are 4.5 cm × 4.5 cm × 0.1 cm. In the real DAC, impurities (e.g., CO, NO2, and SO2) in the air may
potentially have an adverse impact on device performance due to the poisoning effects on Pt-based catalysts. However, the concentration of these impurities is very low (1.4 ppm of CO, 28.7
ppb of NO2, 23.9 ppb of SO2)43; therefore, the effect of impurities is not taken into account in the proof-of-concept device. Transition-metal single-atom catalysts (SACs), such as Co-SAC
catalysts, are promising candidates to prevent the poisoning effects8. CO2 ABSORPTION MODULE The CO2 absorption was performed in a base tank. The base tank has an inner diameter of 14.5 cm.
The air in the laboratory was introduced to the base tank by an air pump. The flow rate of the air pump was quantified by a mass flow meter. Prior to entering the mass flow meter, the air
undergoes a dehydration process through condensation in order to prevent potential damage to the mass flow meter during long periods of operation. The air from the mass flow meter was
introduced into a bubble disk immersed in the KOH solution, which had an approximate effective diameter of 9.0 cm. BPMED MODULE The BPMED module was a home-built single stack cell that
consisted of two compartments: an anode compartment (i.e., base compartment) and a cathode compartment (i.e., acid compartment). The anode and the cathode compartments were separated by a
Fumasep bipolar membrane (FBM-PK). The silicone gaskets were utilized for sealing purposes, specifically between the compartment and end plate, as well as between the compartment and the
BPM. Each compartment has a volume of ∼5.06 mL and a cross-sectional area of 20.25 cm2 (4.5 cm × 4.5 cm). THERMOCHEMICAL METHANATION MODULE The CO2 methanation reaction was performed in a
methanation reactor with a Ru/Al2O3 commercial catalyst. The reactor was a quartz tube with an outer diameter of 8 mm and an inner diameter of 6 mm. Fiberglass wool was filled in the middle
of the reactor. The reactor was then erected, and 0.2 g of Ru/Al2O3 catalyst powder was added. The reactor was then placed horizontally. An alumina ceramic heating tube with an inner
diameter of 10 mm was utilized. The type K thermocouple is positioned between the reactor and the ceramic heating tube. The temperature is manually adjusted based on the temperature feedback
to maintain it within the range of 320 °C. PRODUCT ANALYSIS The product compositions were analyzed using a gas chromatograph (GC). First, the output gas was diluted with pure argon (Ar) gas
in a mixing chamber. Second, the diluted gas was split with a T-junction to input the GC and a solution tank, respectively. The flow rate of the diluted gas into the GC was regulated by
manipulating the liquid level in the solution tank. The gas flow rate was consistent with that of the standard gas used for GC calibration. The product output rates of the hybrid
electro-thermochemical device were calculated by the concentration of product components and the flow rate of dilution gas (i.e., Ar). The carbon removal efficiency (_ξ_removal) was
calculated using the following equation: $${\xi }_{{{\rm{removal}}}}=\frac{{r}_{{{{\rm{CO}}}}_{2}\left({{\rm{g}}}\right)}}{{r}_{{{\rm{DIC}}}}}\times 100\%$$ (10) Where
\({r}_{{{{\rm{CO}}}}_{2}\left({{\rm{g}}}\right)}\) is the CO2 output rate (mol min−1) in the outlet of acid tank and _r_DIC is the DIC input rate (\({\mbox{mol}}\) min−1) in the inlet of
cathode compartment. The current efficiency (_ξ_current) was calculated using the following equation: $${\xi
}_{{{\rm{current}}}}=\frac{{I}_{{{{\rm{CO}}}}_{2}}+{I}_{{{{\rm{H}}}}_{2}}}{{I}_{{{\rm{applied}}}}}\times 100\%$$ (11) Where \({I}_{{{{\rm{CO}}}}_{2}}\) is the current (C s−1) used to CO2
release, \({I}_{{{{\rm{H}}}}_{2}}\) is the current (C s−1) used to H2 production (Supplementary Note 6 the detailed calculations) and _I_applied is the applied current (C s−1). In general,
the Pt catalyst exhibits weaker activity for electrochemical CO2 reduction44,45. Therefore, under room temperature and in acid electrolyte, the potential CO2 reduction products, such as
HCOOH, were ignored. EXPERIMENTAL APPARATUS CO2 sensor (JX-411D-II, Weihai Jingxun Unblocked Electronic Technology Co., LTD). Gas chromatograph (GC9790PLUS, Zhejiang Fuli Analytical
Instruments Co., Ltd.). Intelligent peristaltic pump (LM60B, Nanjing Runze Fluid Control Equipment CO., Ltd.). pH meter (SIN-PH-5018/SIN-PH-5015, Hangzhou Liance Measurement Automation
Technology Co., Ltd.). Paper less recorder (SIN-R200T, Hangzhou Liance Measurement Automation Technology Co., Ltd.). Mass flow meter (CS200, Beijing Sevenstar Flow Co., Ltd.). Source meter
(2601B, Tektronix Company) and DC power supply (MS-1520DS, MAISHENG®). Scanning electrochemical microscope (CHI920D, Shanghai Chenhua Instrument Co., Ltd.). DATA AVAILABILITY The data for
plotting all figures in the manuscript are provided as supplementary data and all other data are available from the corresponding author upon request. Source data are provided with this
paper. REFERENCES * Dlugokencky, E. & Tans, P. Trends in atmospheric carbon dioxide. _NOAA/GML_ _(gml.noaa.gov/ccgg/trends)_. * Hansen, J. et al. Young people’s burden: requirement of
negative CO2 emissions. _Earth Syst. Dynam._ 8, 577–616 (2017). Article ADS Google Scholar * Brethomé, F. M., Williams, N. J., Seipp, C. A., Kidder, M. K. & Custelcean, R. Direct air
capture of CO2 via aqueous-phase absorption and crystalline-phase release using concentrated solar power. _Nat. Energy_ 3, 553–559 (2018). Article ADS Google Scholar * Sanz-Pérez, E. S.,
Murdock, C. R., Didas, S. A. & Jones, C. W. Direct capture of CO2 from ambient air. _Chem. Rev._ 116, 11840–11876 (2016). Article PubMed Google Scholar * Erans, M. et al. Direct air
capture: process technology, techno-economic and socio-political challenges. _Energy Environ. Sci._ 15, 1360–1405 (2022). Article CAS Google Scholar * Eisaman, M. D. et al.
Energy-efficient electrochemical CO2 capture from the atmosphere. _Technical proceedings of the 2009 clean technology conference and trade show_ (2009). * Zhao, R. et al. Thermodynamic
exploration of temperature vacuum swing adsorption for direct air capture of carbon dioxide in buildings. _Energy Convers. Manag._ 183, 418–426 (2019). Article ADS CAS Google Scholar *
Zhu, P. et al. Continuous carbon capture in an electrochemical solid-electrolyte reactor. _Nature_ 618, 959–966 (2023). Article ADS CAS PubMed Google Scholar * Prajapati, A. et al.
Migration-assisted, moisture gradient process for ultrafast, continuous CO2 capture from dilute sources at ambient conditions. _Energy Environ. Sci._ 15, 680–692 (2022). Article CAS Google
Scholar * Pang, S. et al. A phenazine-based high-capacity and high-stability electrochemical CO2 capture cell with coupled electricity storage. _Nat. Energy_ 8, 1126–1136 (2023). * Shu,
Q., Haug, M., Tedesco, M., Kuntke, P. & Hamelers, H. V. M. Direct air capture using electrochemically regenerated anion exchange resins. _Environ. Sci. Technol._ 56, 11559–11566 (2022).
Article ADS CAS PubMed PubMed Central Google Scholar * Shu, Q., Legrand, L., Kuntke, P., Tedesco, M. & Hamelers, H. V. M. Electrochemical regeneration of spent alkaline absorbent
from direct air capture. _Environ. Sci. Technol._ 54, 8990–8998 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Lee, W. H. et al. Sorbent-coated carbon fibers for direct
air capture using electrically driven temperature swing adsorption. _Joule_ 7, 1241–1259 (2023). Article CAS Google Scholar * Keith, D. W., Holmes, G., Angelo, D. S. & Heidel, K. A
process for capturing CO2 from the atmosphere. _Joule_ 2, 1573–1594 (2018). Article CAS Google Scholar * Sgouridis, S., Carbajales-Dale, M., Csala, D., Chiesa, M. & Bardi, U.
Comparative net energy analysis of renewable electricity and carbon capture and storage. _Nat. Energy_ 4, 456–465 (2019). Article ADS CAS Google Scholar * Sharifian, R., Wagterveld, R.
M., Digdaya, I. A., Xiang, C. & Vermaas, D. A. Electrochemical carbon dioxide capture to close the carbon cycle. _Energy Environ. Sci._ 14, 781–814 (2021). Article CAS Google Scholar
* Cooper, C. A technical basis for carbon dioxide storage. _Energy Procedia_ 1, 1727–1733 (2009). Article CAS Google Scholar * Chen, Y. et al. Cooperative catalysis coupling
photo-/photothermal effect to drive Sabatier reaction with unprecedented conversion and selectivity. _Joule_ 5, 3235–3251 (2021). Article CAS Google Scholar * Yan, X. et al.
Nickel@Siloxene catalytic nanosheets for high-performance CO2 methanation. _Nat. Commun._ 10, 2608 (2019). Article ADS PubMed PubMed Central Google Scholar * Lee, M. G. et al. Selective
synthesis of butane from carbon monoxide using cascade electrolysis and thermocatalysis at ambient conditions. _Nat. Catal_. 6, 310–318 (2023). * Welch, A. J. et al. Comparative
technoeconomic analysis of renewable generation of methane using sunlight, water, and carbon dioxide. _ACS Energy Lett._ 6, 1540–1549 (2021). Article CAS Google Scholar * Ding, J. et al.
A tin-based tandem electrocatalyst for CO2 reduction to ethanol with 80% selectivity. _Nat. Energy_ 8, 1386–1394 (2023). Article ADS CAS Google Scholar * Schwach, P., Pan, X. & Bao,
X. Direct conversion of methane to value-added chemicals over heterogeneous catalysts: Challenges and prospects. _Chem. Rev._ 117, 8497–8520 (2017). Article CAS PubMed Google Scholar *
Xu, D., Sullivan, I., Xiang, C. & Lin, M. Comparative study on electrochemical and thermochemical pathways for carbonaceous fuel generation using sunlight and air. _ACS Sustain. Chem.
Eng._ 10, 13945–13954 (2022). Article CAS Google Scholar * Jiang, H. et al. Light-driven CO2 methanation over Au-grafted Ce0.95Ru0.05O2 solid-solution catalysts with activities
approaching the thermodynamic limit. _Nat. Catal_. 6, 519–530 (2023). * Dai, Y. et al. Manipulating local coordination of copper single atom catalyst enables efficient CO2-to-CH4 conversion.
_Nat. Commun._ 14, 3382 (2023). Article ADS CAS PubMed PubMed Central Google Scholar * Obasanjo, C. A. et al. High-rate and selective conversion of CO2 from aqueous solutions to
hydrocarbons. _Nat. Commun._ 14, 3176 (2023). Article ADS CAS PubMed PubMed Central Google Scholar * Toh, W. L., Dinh, H. Q., Chu, A. T., Sauvé, E. R. & Surendranath, Y. The role
of ionic blockades in controlling the efficiency of energy recovery in forward bias bipolar membranes. _Nat. Energy._ 8, 1405–1416 (2023). * Lee, L. & Kim, D. Poly(arylene ether
ketone)-based bipolar membranes for acid–alkaline water electrolysis applications. _J. Mater. Chem. A_ 9, 5485–5496 (2021). Article CAS Google Scholar * Lei, Q., Wang, B., Wang, P. &
Liu, S. Hydrogen generation with acid/alkaline amphoteric water electrolysis. _J. Energy Chem._ 38, 162–169 (2019). Article Google Scholar * Ho, A. et al. Decoupling H2(g) and O2(g)
production in water splitting by a solar-driven V3+/2+(aq,H2SO4)|KOH(aq) Cell. _ACS Energy Lett._ 4, 968–976 (2019). Article CAS Google Scholar * Luo, J. et al. Bipolar membrane-assisted
solar water splitting in optimal pH. _Adv. Energy Mater._ 6, 1600100 (2016). Article ADS Google Scholar * Marin, D. H. et al. Hydrogen production with seawater-resilient bipolar membrane
electrolyzers. _Joule_ 7, 765–781 (2023). Article CAS Google Scholar * Li, D. et al. Highly quaternized polystyrene ionomers for high performance anion exchange membrane water
electrolysers. _Nat. Energy_ 5, 378–385 (2020). Article ADS CAS Google Scholar * Vincent, I. & Bessarabov, D. Low cost hydrogen production by anion exchange membrane electrolysis: A
review. _Renew. Sust. Energ. Rev._ 81, 1690–1704 (2018). Article CAS Google Scholar * Oener, S. Z., Foster, M. J. & Boettcher, S. W. Accelerating water dissociation in bipolar
membranes and for electrocatalysis. _Science_ 369, 1099–1103 (2020). Article ADS CAS PubMed Google Scholar * Digdaya, I. A. et al. A direct coupled electrochemical system for capture
and conversion of CO2 from oceanwater. _Nat. Commun._ 11, 4412 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Gao, J. et al. A thermodynamic analysis of methanation
reactions of carbon oxides for the production of synthetic natural gas. _RSC Adv._ 2, 2358–2368 (2012). Article ADS CAS Google Scholar * Anne J. M. et al. Ph.D. Mars atmospheric
conversion to methane and water: An engineering model of the Sabatier reactor with characterization of Ru/Al2O3 for long duration use on Mars. _47th International Conference on Environmental
Systems 16-20 July 2017, Charleston, South Carolina_ (2017). * Angulo, A., van der Linde, P., Gardeniers, H., Modestino, M. & Fernández Rivas, D. Influence of Bubbles on the Energy
Conversion Efficiency of Electrochemical Reactors. _Joule_ 4, 555–579 (2020). Article CAS Google Scholar * Lu, X. & Zhao, C. Electrodeposition of hierarchically structured
three-dimensional nickel–iron electrodes for efficient oxygen evolution at high current densities. _Nat. Commun._ 6, 6616 (2015). Article ADS CAS PubMed Google Scholar * Yang, F., Chen,
Y., Cheng, G., Chen, S. & Luo, W. Ultrathin nitrogen-doped carbon coated with CoP for efficient hydrogen evolution. _ACS Catal._ 7, 3824–3831 (2017). Article CAS Google Scholar *
Yang, J., Kang, S., Ji, Z., Yin, X. & Tripathee, L. Investigating air pollutant concentrations, impact factors, and emission control strategies in western China by using a regional
climate-chemistry model. _Chemosphere_ 246, 125767 (2020). Article CAS PubMed Google Scholar * Azuma, M., Hashimoto, K., Hiramoto, M., Watanabe, M. & Sakata, T. Carbon dioxide
reduction at low temperature on various metal electrodes. _J. Electroanal. Chem. Interfacial Electrochem._ 260, 441–445 (1989). Article CAS Google Scholar * Hori, Y., Wakebe, H.,
Tsukamoto, T. & Koga, O. Electrocatalytic process of CO selectivity in electrochemical reduction of CO2 at metal electrodes in aqueous media. _Electrochim. Acta_ 39, 1833–1839 (1994).
Article CAS Google Scholar Download references ACKNOWLEDGEMENTS The National Natural Science Foundation of China under grant no. 52376191 and Nathional Key Research and Development
Program of China under grant no. 2023YFB4104600 are acknowledged. The Shenzhen Science and Technology Innovation Commission under Grant No. KCXST20221021111207017, Shenzhen Key Laboratory of
Intelligent Robotics and Flexible Manufacturing Systems under grant no. ZDSYS20220527171403009, Guangdong Basic and Applied Basic Research Foundation under Grant No. 2023A1515011595,
Guangdong Major Project of Basic Research under grant no. 2023B0303000002, SUSTech High Level of Special Funds under grant no. G03034K001, and Guangdong grant under Grant No. 2021QN02L562
are acknowledged. S.D. and Y.H. also acknowledge the National Natural Science Foundation of China under Grant (5247060362) and the Key Project of Natural Science Funds of Tianjin City
(22JCZDJC00540). The computation in this work is supported by Center for Computational Science and Engineering at the Southern University of Science and Technology. AUTHOR INFORMATION
AUTHORS AND AFFILIATIONS * Shenzhen Key Laboratory of Intelligent Robotics and Flexible Manufacturing Systems, Department of Mechanical and Energy Engineering, SUSTech Energy Institute for
Carbon Neutrality, Southern University of Science and Technology, Shenzhen, 518055, China Yaowei Huang, Da Xu & Meng Lin * School of Mechanical Engineering, Tianjin University, Tianjin,
300350, China Yaowei Huang & Shuai Deng * National Industry-Education Platform for Energy Storage, Tianjin University, Tianjin, 300350, China Yaowei Huang & Shuai Deng Authors *
Yaowei Huang View author publications You can also search for this author inPubMed Google Scholar * Da Xu View author publications You can also search for this author inPubMed Google Scholar
* Shuai Deng View author publications You can also search for this author inPubMed Google Scholar * Meng Lin View author publications You can also search for this author inPubMed Google
Scholar CONTRIBUTIONS M.L. and S.D. conceived and supervised the project. Y.H. performed the energy consumption analysis, techno-economic analysis, experiments, data processing and wrote the
draft of the paper. D.X. designed and tested the thermochemical methanation module. All authors contributed to the writing. CORRESPONDING AUTHORS Correspondence to Shuai Deng or Meng Lin.
ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Mahinder Ramdin and Hakhyeon Song for
their contribution to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional
claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA
1 SUPPLEMENTARY DATA 2 SUPPLEMENTARY DATA 3 SUPPLEMENTARY DATA 4 SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission
under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons
licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Huang, Y., Xu, D., Deng, S. _et al._ A hybrid electro-thermochemical device
for methane production from the air. _Nat Commun_ 15, 8935 (2024). https://doi.org/10.1038/s41467-024-53336-9 Download citation * Received: 19 January 2024 * Accepted: 10 October 2024 *
Published: 16 October 2024 * DOI: https://doi.org/10.1038/s41467-024-53336-9 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link
Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative