Pan-cancer whole-genome comparison of primary and metastatic solid tumours
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Metastatic cancer remains an almost inevitably lethal disease1,2,3. A better understanding of disease progression and response to therapies therefore remains of utmost importance.
Here we characterize the genomic differences between early-stage untreated primary tumours and late-stage treated metastatic tumours using a harmonized pan-cancer analysis (or reanalysis) of
two unpaired primary4 and metastatic5 cohorts of 7,108 whole-genome-sequenced tumours. Metastatic tumours in general have a lower intratumour heterogeneity and a conserved karyotype,
displaying only a modest increase in mutations, although frequencies of structural variants are elevated overall. Furthermore, highly variable tumour-specific contributions of mutational
footprints of endogenous (for example, SBS1 and APOBEC) and exogenous mutational processes (for example, platinum treatment) are present. The majority of cancer types had either moderate
genomic differences (for example, lung adenocarcinoma) or highly consistent genomic portraits (for example, ovarian serous carcinoma) when comparing early-stage and late-stage disease.
Breast, prostate, thyroid and kidney renal clear cell carcinomas and pancreatic neuroendocrine tumours are clear exceptions to the rule, displaying an extensive transformation of their
genomic landscape in advanced stages. Exposure to treatment further scars the tumour genome and introduces an evolutionary bottleneck that selects for known therapy-resistant drivers in
approximately half of treated patients. Our data showcase the potential of pan-cancer whole-genome analysis to identify distinctive features of late-stage tumours and provide a valuable
resource to further investigate the biological basis of cancer and resistance to therapies. SIMILAR CONTENT BEING VIEWED BY OTHERS LIMITED EVOLUTION OF THE ACTIONABLE METASTATIC CANCER
GENOME UNDER THERAPEUTIC PRESSURE Article 09 August 2021 WHOLE GENOME SEQUENCING OF METASTATIC COLORECTAL CANCER REVEALS PRIOR TREATMENT EFFECTS AND SPECIFIC METASTASIS FEATURES Article Open
access 25 January 2021 ANALYSIS OF 10,478 CANCER GENOMES IDENTIFIES CANDIDATE DRIVER GENES AND OPPORTUNITIES FOR PRECISION ONCOLOGY Article Open access 18 June 2024 MAIN Metastatic spread
involves tumour cell detachment from a primary tumour, colonization of a secondary tissue and growth in a hostile environment1,2. Advanced metastatic tumours are frequently able to resist
aggressive treatment regimes6. Despite the many efforts to understand these phenomena3,7,8,9, we still have limited knowledge of the contribution of genomic changes that equip tumours with
these extraordinary capacities. Thus, it is essential to characterize genomic differences between primary and metastatic cancers and quantify their effect on therapy resistance to understand
and harness therapeutic interventions that establish more effective and more personalized therapies10. Although extensive whole-genome analyses of primary or metastatic tumour types have
been conducted4,5, large-scale comparative studies between the two tumour stages remain limited due to the logistical challenges associated with obtaining pan-cancer cohorts of primary and
metastatic cancers. To circumvent this issue, most comparison studies have relied on unpaired whole-exome data or have adopted more targeted approaches with a specific focus on driver gene
landscapes11,12,13. However, these efforts have frequently involved separated processing pipelines for primary and metastatic cohorts, complicating the analysis of genomic features that are
highly sensitive to the selected data-processing strategy14,15. A recent study that uniformly analysed more than 25,000 tumours16 has provided a comprehensive overview of the genomic
differences, driver alteration patterns and organotropism using clinical gene-panel sequencing as a base. However, this genomic analysis approach prevented the exploration of the full
spectrum of genomic alterations that have a role in tumorigenesis, such as structural variation and mutational scarring. HARMONIZED WHOLE-GENOME-SEQUENCED TUMOURS Here we created a uniformly
processed whole-genome-sequenced (WGS) inventory of 7,108 matched tumour and normal genomes from two unpaired primary and metastatic cohorts. We first collated the Hartwig Medical
Foundation (Hartwig) dataset5, which included 4,784 samples from 4,375 patients with metastatic cancers. Then, we reprocessed 2,835 primary tumour samples from the Pan-Cancer Analysis of
Whole Genomes (PCAWG) consortium4 using the open-source Hartwig analytical pipeline5,17 to harmonize somatic calling and annotations of events, and to eliminate processing biases (Extended
Data Fig. 1 and Supplementary Table 1). Reassuringly, per-sample comparison of the number of single-base substitutions (SBSs), double-base substitutions (DBSs), indels (IDs) and structural
variants (SVs) revealed a strong agreement between our results and the consensus calls originally generated by the PCAWG consortium (Supplementary Note 1). In addition, our processing
pipeline strategy was minimally affected by differences in sequencing coverage, enabling a reasonable comparison of WGS samples from heterogeneous sources (Supplementary Note 1). A total of
7,108 tumour samples from 71 cancer types met the processing pipeline quality standards (Methods and Extended Data Fig. 1a) and constitutes one of the largest publicly available datasets of
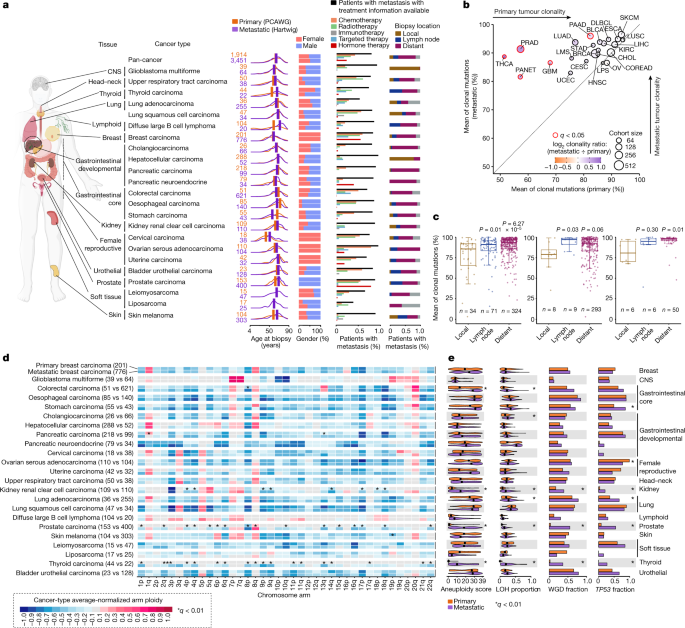
WGS tumours. We next focused on 23 cancer types from 14 tissues with sufficient sample representation, comprising 5,365 tumour samples (1,914 primary and 3,451 metastatic) to explore genomic
differences between primary and metastatic tumours (Fig. 1a and Extended Data Fig. 1a). Within this dataset, patients with metastatic tumours were slightly older at biopsy than patients
with primary tumours (mean of 1.67 years older across all cancer types), although patients with metastatic prostate and thyroid carcinomas, and diffuse large B cell lymphoma were markedly
older than their primary counterparts. Consistent gender proportions were observed across all cancer types except for thyroid adenocarcinomas (metastatic: 72% male and 28% female; primary:
25% male and 75% female). Treatment information was available for 83.7% of patients with metastatic tumours, which is essential to gauge specific treatment-induced contributions to genomic
differences between primary and metastatic tumours (Fig. 1a). Finally, biopsy locations were annotated for 84.2% of patients with metastatic tumours (12.2% from metastatic lesions in the
primary tissue (local), 16.2% in lymph nodes and 55.7% in distant locations) and displayed a highly tumour-type-specific distribution pattern, probably reflecting both the dissemination
patterns of the tumours and the accessibility for safe clinical sampling. COMPARISON OF GLOBAL GENOMIC FEATURES We first explored global genomic differences between primary and metastatic
tumours across the aforementioned 23 cancer types. Metastatic tumours showed an overall increase in clonality compared with their primary tumour counterparts (Fig. 1b). Particularly, five
cancer types had a significantly higher metastatic average clonality ratio, ranging from 13.6% increased mean clonality in pancreatic carcinoma to 37.2% in thyroid carcinoma. Within the
group of patients with metastatic breast carcinoma, distant and lymph node tumour biopsies showed significantly higher clonality ratios than local metastatic lesions (Fig. 1c and
Supplementary Note 2). This increase in clonality was also observed in distant tumour biopsies of oesophageal and colorectal carcinomas (Fig. 1c). Nevertheless, the biopsy location did not
influence tumour clonality in other cancer types such as lung adenocarcinoma and skin melanoma (Extended Data Fig. 1b), suggesting that patterns of tumour dissemination are highly
tumour-type specific9. Our results support the model that metastatic lesions generally have lower intratumour heterogeneity16, which may be explained by a single major subclone seeding event
from the primary cancer and/or by severe evolutionary constraints imposed by anticancer therapies. Comparison of chromosome arm aneuploidy profiles revealed a generally conserved portrait,
which was strongly shaped by the cell of origin (Fig. 1d and Supplementary Table 2), supporting the notion that tumour karyotype is generally defined at early stages of tumorigenesis18. Only
metastatic kidney renal clear cell, prostate and thyroid carcinomas showed substantial changes compared with primary tumours, encompassing 91% (43 of 47) of all significant discrepancies.
Besides the poor prognostic marker 8q gain in metastatic prostate carcinoma19, all discrepancies were associated with an increased prevalence of chromosomal arm losses at the metastatic
setting. Remarkably, 30% (14 of 46) of the metastatic-enriched chromosome arm losses were retained when comparing non-WGD tumours (Extended Data Fig. 1c,d), indicating that other factors,
aside from whole-genome doubling20,21, have an important role in the accumulation of arm-sized chromosomal aberrations. The same three cancer types also showed persistent increases in
specific genomic instability indicators (that is, chromosomal aneuploidy score20, loss of heterozygosity (LOH) genome fraction, WGD22 and _TP53_ alterations23) in the metastatic cohort (Fig.
1e and Supplementary Table 2). Although all of these indicators are elevated pan-cancer in WGD tumours (Extended Data Fig. 1c,d), non-WGD metastatic tumours of these three cancer types also
had significantly greater aneuploidy and LOH scores. Furthermore, patients with metastatic tumours from other cancer types, including lung and colorectal adenocarcinomas, also displayed a
moderate increase in aneuploidy and LOH scores, although they seemed to be primarily associated with higher metastatic WGD rates (Extended Data Fig. 1c). Our results thus revealed that the
majority of cancer types have already acquired variable degrees of chromosomal arm aneuploidy in early stages of tumorigenesis. However, in certain cancers, such as kidney renal clear cell,
prostate and thyroid carcinomas, significantly increased levels of genomic instability were induced in later evolutionary stages, which were, in turn, associated with substantial additional
karyotypic changes. TUMOUR MUTATION BURDEN We observed that the small variant tumour mutation burden (TMB), collectively encompassing SBSs, DBSs and IDs, was only moderately increased in
metastatic tumours compared with primary tumours across the 23 cancer types tested (fold-change increases of 1.25 ± 0.47 for SBSs, 1.55 ± 0.86 for DBSs and 1.45 ± 0.53 for IDs; mean ±
standard deviation (s.d.)). In fact, 15 of the 23 cancer types had no significant increase in mutation burden for any mutation type. Only five cancer types (breast, cervical, thyroid and
prostate carcinomas and pancreatic neuroendocrine tumour) had a consistent increase for the three mutation types at the metastatic stage, although the mutation profiles lacked systematic
differences between primary and metastatic tumours (Fig. 2b and Extended Data Fig. 2a). Finally, further TMB comparisons grouping by tumour subtypes, metastatic biopsy locations and primary
clinical progression status generally provided consistent results, although cancer-type-specific particularities are present (Supplementary Note 2). These results show that TMB is not
necessarily indicative of tumour progression status and that the overall mutational spectra are tightly shaped by the mutational processes that were already active before and during primary
tumour development. MUTATIONAL PROCESSES ACTIVITY COMPARISON To determine whether the TMB differences may be attributed to differential activity of environmental or endogenous mutational
processes, we assessed the activities of all operative mutational signatures in a quantitative and relative manner. We found that mutations attributed to cytotoxic treatments were
significantly enriched in ten cancer types (Fig. 2c (red top bars) and Extended Data Figs. 2b,c and 3a for relative contributions). Platinum-based chemotherapies (SBS31/SBS35 and DBS5)
showed the strongest mutagenic effect with 551 ± 575 (mean ± s.d.) SBS mutations and 32 ± 22 (mean ± s.d.) DBS-attributed mutations on average per sample. In fact, the excess in DBS mutation
burden observed in eight cancer types (breast, oesophageal, stomach, cervical, ovarian serous and lung squamous carcinomas, cholangiocarcinoma and lung adenocarcinoma) was uniquely linked
to platinum treatment mutations (Extended Data Fig. 2b, top bars). Likewise, median mutation contribution from the radiotherapy ID signature24 (ID8) was enriched in six cancer types commonly
exposed to radiation-based treatment (Extended Data Fig. 2c), whereas the 5-fluorouracil25 (SBS17a/b) and polycyclic aromatic hydrocarbon metabolites from chemotreatments26 (DBS2) also
displayed greater metastatic mutation burden contribution in a tumour-type-specific manner (Extended Data Fig. 2b,c). The broad enrichment of SBS2/SBS13 mutations in metastatic cancers
suggests enhanced activity of APOBEC mutagenesis during the progression of advanced tumours. Specifically, our results revealed an increase in APOBEC mutation burden of 325 ± 178 (mean ±
s.d.) mutations per sample in six metastatic tumours (breast, colorectal, stomach, kidney, prostate and pancreatic neuroendocrine carcinomas) that reached statistical significance, with
breast and stomach cancers the types with the strongest increase (more than 500 APOBEC mutations per sample). Other cancer types, such as cervical and bladder urothelial carcinomas, also
showed enhanced APOBEC activity (more than 2,500 median mutations per sample), but they did not reach significance due to already high intrinsic APOBEC activity in the primary tumours. The
metastatic breast cancer samples also had a higher percentage in clustered APOBEC hypermutation variants than primary tumours (15% versus 5%; Extended Data Fig. 2d and Supplementary Table
3). Six metastatic cancer types also displayed more mutations from the clock-like mutational processes, including five cancer types (diffuse large B cell lymphoma, breast, prostate,
pancreatic neuroendocrine and kidney renal clear cell carcinomas) that exhibited an increased SBS1 contribution and three cancer types (hepatocellular, prostate and thyroid carcinomas) that
had an increased SBS5/SBS40 mutation burden. The increase in clock-like mutations in thyroid and prostate cancers, as well as diffuse B cell lymphomas to a lesser extent, may be explained by
a larger proportion of older patients with metastatic disease. However, SBS1 metastatic enrichment was also present in cancer types with highly similar age population distributions (Fig.
1a). Additional focused analyses will be needed to obtain a better understanding of the mutational signature differences that we observed in smaller subsets of cancer types (and subtypes)
and metastatic locations (full data in Supplementary Table 3 and Supplementary Data 1). DIFFERENTIAL SBS1 MUTATION BURDEN To investigate the SBS1 mutation burden differences in more detail,
we evaluated their SBS1 mutation burden by the age of biopsy separately for both cohorts. As expected, the SBS1 mutation burden per year was highly tissue specific27,28 and displayed an
increase with age in the majority of cancer types in both primary and metastatic cohorts (Pearson’s _R_ > 0.1, 15 of 23 tumour types; Extended Data Fig. 4a and Supplementary Table 4).
However, four cancer types (that is, breast, prostate, kidney renal clear cell and thyroid carcinomas) showed an age-independent and significant enrichment of SBS1 mutations in metastatic
lesions (Extended Data Fig. 4a). For instance, metastatic breast cancer had a nearly uniform fold-change increase of 1.46 over primary tumours (188 ± 16 SBS1 mutations, mean ± s.d.) across
the ages of biopsies, and which was generally consistent across breast cancer subtypes (Extended Data Fig. 4b and Supplementary Note 3). This pattern was highly cancer-type specific and was
not observed for most cancer types, including those with similar intratumour heterogeneity in the primary cohort (for example, colorectal, ovarian serous and pancreatic carcinomas) (Extended
Data Fig. 4a). Moreover, this pattern was not explained by differences in tumour genome ploidy (Extended Data Fig. 4c) or by metastatic biopsy location (Supplementary Note 3), was observed
in paired primary–metastatic biopsies from individual patients with breast and kidney renal clear cell carcinomas and rendered consistent patterns when relying on independent unpaired
cohorts (Supplementary Note 3). Finally, other mutational processes that operate over the evolution of the somatic tissues (for example, clock-like mutations attributed to SBS5/SBS40 that
accumulate with age in a cell-cycle-independent manner28,29) did not show such enrichment (Extended Data Fig. 4d). SBS1 mutation burden has been extensively correlated with estimated stem
cell division rates30. Therefore, an increase in age and tumour-type-specific SBS1 mutation burden in treated metastatic tumours may indicate that these tumours have undergone a higher
number of cell divisions. However, the estimated number of years to explain the SBS1 mutation burden shift (23 and 71 years for breast and prostate cancers, respectively; see Supplementary
Table 4) shows that this is unlikely to be the main cause. Hence, a more plausible explanation, which also supports previous observations31,32, is that these metastatic tumours display
accelerated cell division rates compared with their primary tumour counterparts (Extended Data Fig. 4e). Supporting this hypothesis, metastatic tumours also had a lower normalized fraction
of clonal SBS1 mutations (Extended Data Fig. 4f). Of note, this pattern was not observed in cancer types with consistently high SBS1 mutagenic dynamics (Extended Data Fig. 4f) and was
indistinguishable for SBS5/SBS40 mutations (Supplementary Note 3). Finally, we observed a negative association between the yearly rate of SBS1 mutation accumulation in primary tumours (a
proxy of stem cell division rates30) and the estimated fold change of the SBS1 mutation rate in the metastatic cohort (Extended Data Fig. 4g,h). This suggests that tumours with an
intrinsically active turnover rate (for example, colorectal carcinomas) preserve their high proliferation rates, whereas others with lower cell division rates (for example, breast, prostate,
kidney and thyroid carcinomas) may acquire higher proliferation rates during the course of cancer progression. Nevertheless, we cannot rule out the contribution of other
tissue-type-specific or tumour-type-specific mechanisms, such as higher rates of 5-methylcytosine deamination, decreased fidelity to repair these mismatches or higher contribution from other
metastatic-specific mutational processes with overlapping mutational contexts. SV BURDEN Comparison of the total number of SVs per tumour revealed an extensive increase in the metastatic
tumours (fold change of 2.5 ± 1.3, mean ± s.d.). This increase was observed in 13 of 23 (56%) cancer types (Fig. 3a, Extended Data Fig. 5a and Supplementary Table 5), and was not generally
explained by differences in sequencing coverage, tumour clonality (Supplementary Note 1) or cancer-subtype composition (Supplementary Note 2). Moreover, the increased SV burden was also
observed for cancer types lacking substantial changes in genomic instability indicators, such as oesophageal and lung squamous cell carcinomas (Fig. 3a). Finally, we observed an increased SV
burden in prostate and pancreatic neuroendocrine primary tumours that eventually progressed compared with those with relatively better prognosis, which in both cases were in turn lower than
the median values in metastatic tumours (Supplementary Note 2). Overall, compared with TMB, the SV analyses revealed a much more widespread pan-cancer effect, with larger increases per
metastatic cancer type that affected almost every cancer type studied. Small (less than 10 kb) deletions were the most enriched SV types in metastatic tumours (2.7 ± 1.2 fold change in 15 of
23 cancer types with significant enrichment, mean ± s.d.; Extended Data Fig. 5a,b). Larger (10 kb or larger) deletions and duplications had a similar pan-cancer enrichment, although
generally with slightly lower fold changes. Complex SVs with 20 or more breakpoints, encompassing events such as chromothripsis and chromoplexy, were enriched in metastatic prostate
carcinomas (more than threefold enrichment). Finally, a strong cancer-type-specific metastatic enrichment was also noted for long interspersed nuclear element insertions (LINE), with an
increased fold change of 12.2 and 12.5 in stomach and bladder urothelial carcinomas, respectively. We next used linear regression models to unravel the underlying features associated with
the observed increase in SV burden in metastatic tumours (Extended Data Fig. 6a–k and Supplementary Table 5). Our approach confirmed the role of previously described cancer-type-specific
driver-induced SV phenotypes, including homologous recombination deficiency33 in metastatic breast carcinoma tumours, _CDK12_ (ref. 34) alterations in prostate carcinoma and _MDM2_ (ref. 35)
amplifications in breast ER+/HER2− carcinomas, among others. Genomic instability features, such as genome ploidy and _TP53_ alterations, showed a strong pan-cancer association with
deletions and duplications (Extended Data Fig. 6a,d), and thus very likely contributed to the observed SV increase in metastatic tumours20,22,23. Finally, previous exposure to radiotherapy
treatment was strongly associated with small deletions in breast ER+/HER2− and prostate carcinomas36. CANCER DRIVER GENE LANDSCAPE Metastatic tumours showed a moderate increase in the total
number of driver gene alterations per patient (a mean of 4.5 and 5.3 driver alterations per sample in primary and metastatic tumours, respectively), including 8 (34%) tumour types with a
significant increase (Fig. 4a). Prostate carcinoma (average increase of 3.16 driver alterations per sample), pancreatic neuroendocrine tumour (2.16), thyroid carcinoma (1.7) and kidney renal
clear cell carcinoma (1.87) showed the strongest increases (more than 1.5 driver alterations per patient), whereas the majority of cancer types showed a mean increase below 1.5 driver
alterations per sample. All mutation types (amplifications, deletions and mutations) contributed to the increased driver alterations in metastatic tumours (Extended Data Fig. 7a). Comparison
of gene and cancer-type frequencies revealed that only 12 genes had a significant frequency bias in at least one cancer type (22 gene and cancer-type pairs in total; Fig. 4b, Extended Data
Fig. 7b and Supplementary Table 6). The majority (19 out of 22, 86%) of the significant pairs had enrichment towards higher metastatic frequency, including four driver genes that were
exclusively mutated in metastatic tumours and were not found in the primary tumour equivalents (_PTPRD_ in kidney renal clear cell carcinoma, _CREBBP_ in pancreatic neuroendocrine tumour,
and _RET_ and _TP53_ alterations in thyroid carcinoma). Most metastatic-enriched cancer drivers had a cancer-type-specific enrichment, including well-established resistance gene drivers
associated with anticancer therapies, such as _AR_ and _ESR1_ alterations in patients with prostate and ER+ breast carcinomas treated with hormone deprivation therapies37,38 (Fig. 4b and
Supplementary Note 2). Nevertheless, three driver genes (that is, _TP53_, _CDKN2A_ and _TERT_) showed a metastatic enrichment across multiple cancer types (Fig. 4b), indicating that
alterations of these genes may enhance aggressiveness by disturbing pan-cancer hallmarks of tumorigenesis. We next investigated whether the reported driver differences may have an effect on
potential clinical actionability. Cancer-type-specific comparison of therapeutically actionable variants revealed an overall larger fraction of patients with therapeutically actionable
variants in the metastatic cohort, with high variability across cancer types (Extended Data Fig. 8a and Supplementary Table 7). Subsetting by A-on label variants (that is, approved
biomarkers in the specific cancer type) revealed a consistent pattern in which only cholangiocarcinoma (_FGFR2_ fusions and _IDH1_ mutations) and lung adenocarcinoma (_EGFR_ alterations)
showed a substantial proportional increase in the metastatic cohort (Extended Data Fig. 8a,b). Non-A-on label biomarkers (A-off label, B-on label and B-off label) showed a modest and
tumour-type-dependent metastatic increase, which was mainly linked to the increased alteration frequency of _KRAS_ exon 2 mutations and _CDKN2A_ loss in advanced tumour stages (Extended Data
Fig. 8b). TREATMENT-ASSOCIATED DRIVERS The presence of treatment resistance driver genes in late-stage tumours prompted us to devise a test that aimed to identify treatment-enriched drivers
(TEDs) that were either significantly enriched (that is, treatment enriched) or exclusively found (that is, treatment exclusive) in a cancer-type-specific and treatment-specific manner
(Extended Data Fig. 9a and Methods). Our analytical framework provided 61 TEDs associated with 33 treatment groups from 8 cancer types and 4 cancer subtypes (Fig. 5a,b, Supplementary Table 8
and Supplementary Note 4). Of the identified TEDs, 33 of 61 (54%) were coding mutation drivers, 16 (26%) were copy number amplifications, 9 (14%) were non-coding drivers and 3 (6%) were
recurrent homozygous deletions (Fig. 5b and Extended Data Fig. 9b,c). Reassuringly, the majority of the top hits were known treatment resistance drivers, including _AR_-activating mutations
and gene amplifications in patients with prostate cancer treated with androgen-deprivation therapy38 (Extended Data Fig. 9d,e), _ESR1_536–538 mutations in patients with breast cancer treated
with aromatase inhibitors37 (Extended Data Fig. 9f), and _EGFR__T790M_ mutations (Extended Data Fig. 9g) and _EGFR_ copy number gains in patients with lung adenocarcinoma treated with
_EGFR_ inhibitors39,40 (Extended Data Fig. 9h), among others. Moreover, we also found that _TP53_ alterations were recurrently associated with resistance to multiple treatments, which may
indicate that these alterations are prognostic markers for enhanced tumour aggressiveness and plasticity rather than being a cancer-type-specific mechanism of drug resistance (Supplementary
Note 4). Our results also provided a long tail of candidate drivers of resistance, some of them with orthogonal evidence by independent reports (Fig. 5b and Extended Data Fig. 9b,c).
Examples of the latter group include _TYMS_ amplification in patients with breast cancer treated with pyrimidine antagonists41 (Extended Data Fig. 9i), _PRNC1_ and _MYC_ co-amplifications in
patients with prostate cancer treated with androgen deprivation42 (Extended Data Fig. 9j), _ACTL6A_ promoter mutations in patients with triple-negative breast cancer treated with
platinum-based therapies43, and _FGFR2_ promoter mutations in patients with breast cancer treated with CDK4/CDK6 inhibitors44. The full TEDs catalogue is provided in Supplementary Table 8
and constitutes a valuable resource for investigating resistance mechanisms to common cancer therapies. Overall, 53% of patients with metastatic disease with annotated treatment information
had TEDs, including 32% with annotations of known resistance drivers and an additional 21% of patients with candidate resistance drivers derived from our analysis (Fig. 5c). We identified
0.70 ± 0.53 (mean ± s.d.) TEDs per metastatic sample across the 8 cancer types that had reported TEDs (Fig. 5d), with prostate and breast carcinomas displaying the greatest prevalence of
TEDs (that is, 1.74 and 1.12 drivers per patient with prostate and breast cancers, respectively). Therefore, after excluding TEDs, primary and metastatic tumours had a 36% reduction of their
original differences in the number of drivers per sample (from 5.3 to 5.0 mean drivers per sample in the metastatic cohort after excluding TEDs, compared with 4.5 mean drivers per sample in
the primary cohort) (Fig. 5d and Supplementary Table 8), indicating that an important proportion of the metastatic-enriched drivers are probably associated with resistance to anticancer
therapies. DISCUSSION In this study, we describe a cohort of more than 7,000 uniformly reprocessed WGS samples from patients with primary untreated and metastatic treated tumours. We
compared genomic features across 23 cancer types and confirmed previous cancer-type-specific observations while also providing novel biological findings, such as the clock-like molecular
features, the prevalence of SV burden across different tumorigenic stages and the incidence of TEDs in treated patients. Specifically, metastatic tumours displayed high genomic instability,
low intratumour heterogeneity and strong accumulation of SVs. However, the magnitude of genomic differences between primary and metastatic tumours was highly cancer-type specific and was
influenced by the exposure to cancer treatments. Overall, five cancer types (prostate, thyroid, kidney renal clear cell, breast and pancreatic neuroendocrine carcinomas) showed an intense
transformation of the genomic landscape in advanced tumorigenic stages (Extended Data Fig. 10, labelled as strong). The other cancer types displayed variable genomic differences, although
the chromosomal genomic portrait tended to be conserved. Cancer types with the strongest genomic differences between primary and metastatic settings in our analyses typically have a good
prognosis in the primary setting. But then, whether the metastatic tumours representing a unique set of primary patients that eventually progressed (that is primaries from the metastatic
cohort were ‘born to be bad’) or whether there are stochastic triggers of metastatic disease in relatively indolent primaries are still to be determined. To fully address these, larger
pan-cancer sets of matched biopsies from the same patient, as already implemented in various cancer-type-specific studies45,46, would be needed. This study faced various limitations, such as
the use of different laboratory workups and sequencing parameters used for primary and metastatic tumour samples, although we demonstrated that this does not severely have an effect on the
overall detectability of clonal somatic variants (Supplementary Note 1). However, we cannot exclude the possibility of missing highly subclonal driver mutations. Furthermore, our
observations are unlikely to be exhaustive, especially for lower frequency events, because of limited cohort (or subcohort) sizes. Expanding cancer cohorts in research or clinical settings
will be essential to advance our understanding of tumour progression. Finally, genomic changes alone cannot entirely explain how tumour cells are able to colonize other organs. Therefore,
additional information from complementary tumour omics47 and from the tumour microenvironment48 will be needed to further dissect and better understand metastasis and resistance to cancer
therapies. To conclude, our dataset constitutes a valuable resource that can be leveraged to further study other aspects of tumour evolution, such as genomic differences across metastatic
biopsy locations (Supplementary Note 2), dedicated analysis for cancer subtypes (Supplementary Note 2), genetic immune escape alterations in primary and metastatic tumours49 as well as for
the development of machine learning tools to foster cancer diagnostics50. METHODS COHORT GATHERING AND PROCESSING We have matched tumour–normal WGS data from patients with cancer from two
independent cohorts: Hartwig and PCAWG. A detailed description of the Hartwig and PCAWG cohort gathering and processing as well as comprehensive documentation of the PCAWG sample reanalysis
with the Hartwig somatic pipeline is described in the Supplementary Note 1. TUMOUR CLONALITY ANALYSIS Each mutation in the .vcf files was given a subclonal likelihood by PURPLE. Following
PURPLE guidelines, we considered mutations with subclonal scores equal or higher than 0.8 to be subclonal and mutations below the 0.8 threshold to be clonal. For each sample, we then
computed the average proportion of clonal mutations by dividing the number of clonal mutations by the total mutation burden (including SBS, multinucleotide variants and IDs). Finally, for
each cancer type, we used Mann–Whitney test to assess the significance of the clonality difference between the primary and metastatic tumours. _P_ values were adjusted for false discovery
rate (FDR) using the Benjamini–Hochberg procedure. An adjusted _P_ < 0.05 was deemed as significant. In addition, we leveraged biopsy site data in patient reports to further investigate
differences in metastatic tumour clonality according to the metastatic biopsy site (see also Supplementary Note 2). If the metastatic biopsy site was in the same organ or tissue as the
primary tumour, we considered them as ‘local’, whereas if the metastatic biopsy site was reported in the lymphoid system or other organs or tissues, they were dubbed as ‘lymph’ and
‘distant’, respectively. Cancer types for which there was a minimum of five samples available for each of the biopsy groups were selected and Mann–Whitney test was used to compare the
clonality between the biopsy groups. KARYOTYPE Chromosome arm level and genome ploidy was estimated as previously described20. First, for each chromosome arm, tumour purity and
ploidy-adjusted copy number (CN) segments (as determined by PURPLE) were rounded to the nearest integer. Second, arm coverage of each integer CN was calculated as the fraction of chromosome
arm bases with the specific CN divided by the chromosome arm length (for example, 60% of all chromosome 5p segments have a CN of 2, 30% have a CN of 1 and 10% have a CN of 3). We defined the
arm-level ploidy level as the CN with the highest coverage across the whole arm (in the example above it would be 2). Third, we computed the most recurrent chromosome arm ploidy levels
across all chromosome arms per sample (that is, observed genome ploidy). Next, we estimated the true genome ploidy by taking WGD status (given by PURPLE) into account. If a sample did not
undergo WGD, its total expected genome ploidy was deemed to be 2_n_. If a sample did undergo WGD and its observed genome ploidy was less than six, the estimated genome ploidy was deemed to
be 4_n_, and 8_n_ if the observed genome ploidy was six or more. An observed genome CN of more than eight was not found in our dataset. Then, for each chromosome arm in each sample, we
defined the normalized arm ploidy as the difference between the arm-level ploidy level and the expected genome ploidy. The resulting value was classified as 1 for differences higher than or
equal to 1 (representing arm gains), as −1 for differences lower than or equal to −1 (representing arm losses) or as 0 (no difference). Normalized arm ploidy values were averaged across all
samples from a cancer type in a cohort-specific manner (that is, separating primary and metastatic samples). A Mann–Whitney test was performed per cancer type and chromosome arm to assess
the mean difference in arm gains or losses at the cancer-type level. The resulting _P_ value was FDR adjusted across all arms per cancer type. Finally, _q_ < 0.01 and a normalized arm
ploidy difference higher than 0.25 was deemed to be significant. GENOMIC INSTABILITY INDICATORS To compare the differences in aneuploidy scores and the LOH proportions in each group, a
Mann–Whitney test was performed per cancer type. The aneuploidy score represents the number of arms per tumour sample that deviate from the estimated genome ploidy as previously described20.
The LOH score of a given sample represents the sum of all LOH regions divided by the GRCh37 total genome length. A genomic region is defined as LOH when the minor allele CN < 0.25 and
major allele CN ≥ 0.8. To compare the fraction of samples with a driver mutation in _TP53_ as well as the fraction of WGD samples per cohort, a Fisher’s exact test was performed per cancer
type. Any _TP53_ driver alteration (non-synonymous mutation, biallelic deletion and homozygous disruption) was considered in the analysis. Multiple driver mutations per sample in a single
gene were considered as one driver event. WGD was defined as present if the sample had more than 10 autosomes with an estimated chromosome CN of more than 1.5. _P_ values were FDR corrected
across all cancer types. A _q_ < 0.01 was deemed to be significant for all statistical tests. MUTATIONAL SIGNATURE ANALYSIS SIGNATURE EXTRACTION The number of somatic mutations falling
into the 96 SBS, 78 DBS and 83 ID contexts (as described in the COSMIC catalogue51; https://cancer.sanger.ac.uk/signatures/) was determined using the R package mutSigExtractor
(https://github.com/UMCUGenetics/mutSigExtractor, v1.23). SigProfilerExtractor (v1.1.1) was then used (with default settings) to extract a maximum of 21 SBS, 8 DBS and 10 ID de novo
mutational signatures. This was performed separately for each of the 20 tissue types that had at least 30 patients in the entire dataset (aggregating primary and metastatic samples; see
Supplementary Table 3). Tissue types with less than 30 patients as well as patients with metastatic tumours with unknown primary location type were combined into an additional ‘Other’ group,
resulting in a total of 21 tissue-type groups for signature extraction. To select the optimum rank (that is, the eventual number of signatures) for each tissue type and mutation type, we
manually inspected the average stability and mean sample cosine similarity plot output by SigProfilerExtractor. This resulted in 440 de novo signature profiles extracted across the 21
tissue-type groups (Supplementary Table 3). Least squares fitting was then performed (using the fitToSignatures() function from mutSigExtractor) to determine the per-sample contributions to
each tissue-type-specific de novo signature. AETIOLOGY ASSIGNMENT The extracted de novo mutational signatures with high cosine similarity (≥0.85) to any reference COSMIC mutational
signatures with known cancer-type associations51 were labelled accordingly (288 de novo signatures matched to 57 COSMIC reference signatures). For the remaining 152 unlabelled de novo
signatures, we reasoned that there could be one or more signatures from one cancer type that is highly similar to those found in other tissue types, and that these probably represent the
same underlying mutational process. We therefore performed clustering to group likely equivalent signatures. Specifically, the following steps were performed: * (1) We calculated the
pairwise cosine distance between each of the de novo signature profiles. * (2) We performed hierarchical clustering and used the base R function cutree() to group signature profiles over the
range of all possible cluster sizes (minimum number of clusters = 2; maximum number of clusters = number of signature profiles for the respective mutation type). * (3) We calculated the
silhouette score at each cluster size to determine the optimum number of clusters. * (4) We grouped the signature profiles according to the optimum number of clusters. This yielded 27 SBS, 7
DBS and 8 ID de novo signature clusters (see Supplementary Table 3). For certain de novo signature clusters, we could manually assign the potential aetiology based on their resemblance to
signatures with known aetiology described in COSMIC51, Kucab et al.26 and Signal (access date 1 February 2023)52. Some clusters were an aggregate of two known signatures, such as
SBS_denovo_clust_2, which was a combination of SBS2 and SBS13, both linked to APOBEC mutagenesis. Other clusters had characteristic peaks of known signatures, such as DBS_denovo_clust_4,
which resembled DBS5 based on having distinctive CT>AA and CT>AC peaks. Finally, DBS_denovo_clust_1 was annotated as a suspected _POLE_ mutation and MMRd, as samples with high
contribution (more than 150 mutations) of this cluster are frequently microsatellite instable (MSI) or have _POLE_ mutations. Likewise, DBS_denovo_clust_2 was annotated as a suspected MMRd
as the aetiology, as samples with high contribution (more than 250 mutations) of this cluster were all MSI. See Supplementary Table 3 for a list of all the manually assigned aetiologies.
After applying the aetiology assignment, the de novo extraction resulted in 69 SBS, 13 DBS and 18 ID representative mutational signatures (Supplementary Table 3). Most of these (42 of 69
SBSs, 7 of 13 DBSs and 8 of 18 IDs) mapped onto the well-described mutational signatures in human cancer35,53. COMPARING THE PREVALENCE OF MUTATIONAL PROCESSES BETWEEN PRIMARY AND METASTATIC
CANCER We then compared the activity (that is, the number of mutations contributing to) of each mutational process between primary and metastatic tumours. For each sample, we first summed
the contributions of signatures of the same mutation type (that is, SBS, DBS or ID) with the same aetiology, hereafter referred to as ‘aetiology contribution’. Per cancer type and per
aetiology, we performed two-sided Mann–Whitney tests to determine whether there was a significant difference in aetiology contribution of primary and metastatic tumours. Per cancer type and
per mutation type, we used the p.adjust() base R function to perform multiple testing correction using Holm’s method. Next, we added a pseudocount of 1 to the contributions (to avoid
dividing by 0) and calculated the median contribution log2 fold change, that is, log2((median contribution in metastatic tumours + 1)/(median contribution in primary tumours + 1)). We
considered the aetiology contribution between primary and metastatic tumours to be significantly different when _q_ < 0.05, and log2 fold change ≥ 0.4 or log2 fold change ≤ −0.4 (= ±
×1.4). RELATIVE CONTRIBUTION Relative aetiology contribution was calculated by dividing aetiology contribution by the total contribution of the respective mutation type (that is, SBS, DBS or
ID). To determine the significant difference in relative aetiology contribution, we performed two-sided Mann–Whitney tests as described above. We also calculated the median difference in
contribution (that is, median relative contribution in metastatic tumours − median relative contribution in primary tumours). We considered the relative aetiology contribution between
primary and metastatic tumours to be significantly different when _q_ < 0.05 and median difference was 0.01 or more. We also determined whether there was an increase in the number of
samples with high aetiology contribution (that is, hypermutators) in metastatic versus primary cohorts. For each signature, a sample was considered a hypermutator if the aetiology
contribution was 10,000 or more for SBS signatures, 500 or more for DBS signatures or 1,000 or more for ID signatures. For each cancer type, for each aetiology, we performed pairwise testing
only for cases in which there were five or more hypermutator samples for either metastatic or primary tumours. Each pairwise test involved calculating _P_ values using two-sided Fisher’s
exact tests, and effect sizes by multiplying Cramer’s _V_ by the sign of the log2(odds ratio) to calculate a signed Cramer’s _V_ value that ranges from −1 to +1 (indicating enrichment in
primary or metastatic, respectively). We then used the p.adjust() base R function to perform multiple testing correction using Bonferroni’s method. SBS1–AGE CORRELATIONS IN PRIMARY AND
METASTATIC TUMOURS To count the SBS1 mutations, we relied on the definition from ref. 54 that is based on the characteristic peaks of the COSMIC SBS1 signature profile: single-base CpG >
TpG mutations in NpCpG context. To ensure that these counts and the downstream analyses are not affected by differential APOBEC exposure in primary and metastatic cohorts, we excluded CpG
> TpG in TpCpG, which is also a characteristic peak in the COSMIC SBS2 signature profile. In addition, for skin melanoma, CpG > TpG in [C/T]pCpG, which overlaps with SBS7a, was
excluded. To obtain the SBS5 and SBS40 counts, we relied on their exposures derived from the mutational signature analyses performed in this study (described above). To assess the
correlation between SBS1 burden and the age of the patient, at biopsy we performed a cancer-type and cohort-specific linear regression (that is, separate regression for primary and
metastatic tumour samples). To avoid spurious effects caused by hypermutated tumours, samples with a TMB greater than 30,000 as well as those with SBS1 burden greater than 5,000 were
excluded. For each cancer type and cohort, we then computed 100 independent linear regressions by randomly selecting 75% of the available samples. We selected the median linear regression
(based on the regression slope) as representative regression for further analyses. Similarly, confidence intervals were derived from the 1st and 99th percentile of the computed regressions.
To evaluate the significance of the differences between primary and metastatic representative linear regressions (hereafter referred to as linear regression for simplicity), we first
filtered out cancer types that failed to show a positive correlation trend between SBS1 burden and age at biopsy in both primary and metastatic tumours (that is, Pearson’s correlation
coefficient of primary and metastatic regression greater than 0.1). Next, for each selected cancer type, we computed the regression residuals of primary and metastatic SBS1 mutation counts
using, in both cases, the primary linear regression as baseline. The primary and metastatic residual distributions were then compared using a Mann–Whitney test to evaluate significance.
Cancer types with a Mann–Whitney _P_ < 0.01 were deemed as significant. Finally, to ensure that the differences were uniform across different age ranges (that is, not driven by a small
subset of patients), we only considered significant cancer types in which the metastatic linear regression intercept was higher than the primary intercept. SBS5/SBS40 correlations were
computed following the same procedure and using the sum of SBS5 and SBS40 exposures for each tumour sample. If none of the mutations were attributed to SBS5/SBS40 mutational signatures, the
aggregated value was set to zero. In the ploidy-corrected analyses, we divided the SBS1 mutation counts (and SBS5/SBS40 mutation counts for the SBS5/SBS40 ploidy-corrected regression,
respectively) by the PURPLE-estimated tumour genome ploidy. For each cancer type, the mean fold change (fc) was defined as \(\underline{{\rm{fc}}}=\frac{1}{40}{\sum
}_{i=40}^{80}\frac{{{\rm{M}}{\rm{Pred}}}_{i}}{{{\rm{P}}{\rm{Pred}}}_{i}}\) where MPred_i_ and PPred_i_ are the estimated number of SBS1 mutations for a given age _i_th according to the
metastatic and primary linear regressions, respectively. Similarly, the mean estimated SBS1 burden difference (SBS1diff) was defined as: \(\underline{{\rm{SBS1diff}}}=\frac{1}{40}{\sum
}_{i=40}^{80}{{\rm{MPred}}}_{i}-{{\rm{PPred}}}_{i}\). CLONALITY OF CLOCK-LIKE MUTATIONS SBS1 individual mutations were identified as described in the previous section. For SBS5 and SBS40
mutations, we used a maximum likelihood approach to assign individual mutations to the SBS5 and SBS40 mutational signatures in a cancer-type-specific manner. For every SBS1 (and SBS5/SBS40
mutation), we then assign the clonality according to the PURPLE subclonal likelihood estimation, in which only mutations with subclonal (SUBCL) likelihood ≥ 0.8 were considered as such (see
above). For each tumour sample, the SBS1 clonality ratio (or respectively SBS5/SBS40 clonality ratio) was defined as the ratio between the proportion of clonal SBS1 mutations
(\(\frac{{\rm{SBS\; 1\; clonal\; mutations}}}{{\rm{SBS\; 1\; mutations}}}\)) divided by the total proportion of clonal alterations in the sample (\(\frac{{\rm{Total\; clonal\;
mutations}}}{{\rm{Total\; mutations}}}\)). PRIMARY SBS1 MUTATION RATE AND METASTATIC SBS1 AGE-CORRECTED ENRICHMENT We computed for each primary cancer type the average number of SBS1 per
year as the number of SBS1 mutations divided by the age of the patient at biopsy (only considering primary samples and excluding hypermutated samples as described above). We then used a
Spearman’s correlation to assess its association with the estimated mean SBS1 mutation rate fold change in metastatic tumours (see above). In addition, to exclude potential biases in our
primary cohort, we repeated the same analysis relying on an independent measurement of primary cancer SBS1 yearly accumulation. Specifically, we used the best-estimated accumulation of SBS1
per year from ref. 30 (Supplementary Table 6) and regressed it to the fold-change estimates for the matching cancer types present in both datasets. SV ANALYSIS DEFINITIONS OF SV TYPE LINX55
chains one or more SVs and classifies these SV clusters into various event types (‘ResolvedType’). We defined deletions and duplications as clusters with a ResolvedType of ‘DEL’ or ‘DUP’
whose start and end breakpoints are on the same chromosome (that is, intrachromosomal). Deletions and duplications were split into those less than 10 kb and 10 kb or more in length (small
and large, respectively), based on observing bimodal distributions in these lengths across cancer types (Extended Data Fig. 5b). We defined complex SVs as clusters with a ‘COMPLEX’
ResolvedType, an inversion ResolvedType (including: INV, FB_INV_PAIR, RECIP_INV, RECIP_INV_DEL_DUP and RECIP_INV_DUPS) or a translocation ResolvedType (including: RECIP_TRANS,
RECIP_TRANS_DEL_DUP, RECIP_TRANS_DUPS, UNBAL_TRANS and UNBAL_TRANS_TI). Complex SVs were split into those with less than 20 and 20 or more SVs (small and large, respectively), based on
observing similar unimodal distributions in the number SVs across cancer types whose tail begins at approximately 20 breakpoints (Extended Data Fig. 5b). Finally, we defined long
interspersed nuclear element insertions (LINEs) as clusters with a ResolvedType of ‘LINE’. For each sample, we counted the occurrence (that is, SV burden) of each of the seven SV types
described above. In addition, we determined the total SV burden by summing counts of the SV types. COMPARING SV BURDEN BETWEEN PRIMARY VERSUS METASTATIC CANCER We then compared the SV-type
burden between primary versus metastatic tumours as shown in Fig. 3a. First, we performed two-sided Mann–Whitney tests per SV type and per cancer type to determine whether there was a
statistically significant difference in SV-type burden between primary versus metastatic. The Bonferroni method was used for multiple testing correction on the _P_ values from the
Mann–Whitney tests (to obtain _q_ values). Next, we calculated relative enrichment as follows: log10(median SV-type burden in metastatic tumours + 1) − log10(median SV-type burden in primary
tumours + 1); and calculated fold change as follows: (median SV-type burden in metastatic tumours + 1) / (median SV-type burden in primary tumours + 1). When calculating relative enrichment
and fold change, the pseudocount of 1 was added to avoid the log(0) and divide by zero errors, respectively. Fold changes are displayed with a ‘>’ in Fig. 3a when the SV burden for
primary tumours is 0 (that is, when a divide by 0 would occur without the pseudocount). We considered the SV-type burden between primary versus metastatic to be significant when: _q_ <
0.05, and fold change ≥ 1.2 or fold change ≤ 0.8 IDENTIFYING FEATURES ASSOCIATED WITH INCREASED SV BURDEN IN METASTATIC CANCER To identify the features that could explain increased SV
burden, we correlated SV burden with various tumour genomic features. This included: (1) genome ploidy (determined by PURPLE); (2) homologous recombination deficiency (determined by CHORD33)
and MSI (determined by PURPLE) status; (3) the presence of mutations in 345 cancer-associated genes (excluding fragile site genes that are often affected by CN alterations5), hereafter
referred to as ‘gene status’; and (4) treatment history, including the presence of radiotherapy, the presence of one of the 79 different cancer therapies as well as the total number of
treatments received. All primary samples and all metastatic samples without treatment information were considered to have no treatment. Genome ploidy and total number of treatments received
were numeric features, whereas all of the remaining were boolean (that is, true or false) features. In total, there were 429 features. SV-type burden was transformed to log10(SV-type burden
+ 1) and was correlated with the 429 features using multivariate linear regression models (LMs). This was performed separately for each of the seven SV types, and for each cancer type (or
subtype). In the SV main analysis (Fig. 4b–f), there were 23 cancer types, resulting in a total of 161 (23 cancer types × 7 SV types) LM models. Each LM model (that is, per SV type and
cancer type) involved training of three independent LMs with (1) both metastatic and primary samples (primary + metastatic), (2) only Hartwig samples (metastatic only), and (3) only PCAWG
samples (primary only). This was done to filter out correlations between features and increased SV-type burden solely due to differences in feature values between primary and metastatic
tumours. We then required features that positively correlated with SV-type burden in the primary + metastatic LM to independently show the same association in the metastatic-only or
primary-only LMs. Only genomic features that independently showed positive correlation with the SV burden were further considered as significant (that is, represented in the lollipop plots).
Each of the three LMs was trained as follows: * (1) Remove boolean features with too few ‘true’ samples. * (i) For the primary + metastatic LM, remove gene status features with less than 15
‘true’ samples. * (ii) For the metastatic-only and primary-only LMs, remove gene status features with less than 10 ‘true’ samples. * (iii) For the remaining boolean features, remove
features with less than 5% ‘true’ samples. * (2) Fit a LM using the lm() base R function to correlate log10(SV-type burden + 1) versus all features. For each LM analysis, we used the
following filtering criteria to identify the features that were correlated with increased SV-type burden: * (1) Only keep LM analyses for which there was significant increase in SV-type
burden for the respective cancer type (_P_ < 0.01 as described in the previous section ‘Comparing SV burden between primary versus metastatic cancer’). * (2) For primary + metastatic LM:
* (i) Regression _P_ < 0.01 * (ii) Coefficient _P_ < 0.01 * (iii) Coefficient more than 0 * (3) For metastatic-only LM or primary-only LM: * (i) Coefficient _P_ < 0.01 * (ii)
Coefficient more than 0 Finally, to determine which features (of those correlated with increased SV-type burden) were enriched in metastatic tumours compared with primary tumours (and vice
versa), we calculated Cliff’s delta for numeric features and Cramer’s _V_ for boolean features. Cliff’s delta ranges from −1 to +1, with −1 representing complete enrichment in primary
tumours, whereas +1 represents complete enrichment in metastatic tumours. Cramer’s _V_ only ranges from 0 to 1 (with 1 representing enrichment in either primary or metastatic tumours), the
sign of the log(odds ratio) was assigned as the sign of the Cramer’s _V_ value so that it ranged from −1 to +1. Features with an effect size of more than 0 were considered as those that
could explain the SV burden increase in metastatic cancer when compared with primary cancer. DRIVER ALTERATIONS We relied on patient-specific cancer driver and fusion catalogues constructed
by PURPLE5 and LINX55. Only drivers with a driver likelihood of more than 0.5 were retained. Fusion drivers were filtered for those that were previously reported in the literature.
Similarly, we manually curated the list of drivers and removed _SMAD3_ hotspot mutations because of the high-burden mutations in low-mappability regions. The final driver catalogue contained
a total of 453 driver genes and the final fusion catalogue contained 554 reported fusions. To compare the number of drivers in primary and metastatic tumours, we then combined fusions with
the LINX driver variants to calculate a patient-specific number of driver events. Drivers that concern the same driver gene but a different driver type were deemed to be single drivers (for
example, _TP53_ mutation and _TP53_ deletion in the same sample were considered as one driver event). Cancer-type-specific Mann–Whitney test was performed to assess differences between
primary and metastatic tumours. An adjusted _q_ < 0.01 was deemed to be significant. To assess the driver enrichment, a contingency matrix was constructed from the driver catalogue,
containing the frequency of driver mutations per driver type (that is, deletion, amplification or mutations) and cancer type in each cohort (metastatic and primary). A second contingency
matrix was constructed for the fusions. Partial amplifications were considered as amplifications, whereas homologous disruptions were considered as deletions. These contingency matrices were
filtered for genes that show a minimum frequency of five mutated samples in either the primary or the metastatic cohorts. Then, a two-sided Fisher’s exact test for each gene, cancer type
and mutation type was performed and the _P_ value was adjusted for FDR per cancer type. Cramer’s _V_ and the odds ratio were used as effect size measures. An adjusted _P_ < 0.01 was
deemed to be significant. THERAPEUTIC ACTIONABILITY OF VARIANTS To determine the amount of actionable variants observed in each sample, we compared our variants annotated by SnpEff (v5.1)56
to those derived from three different databases (OncoKB57, CIViC58 and CGI59) that were classified based on a common clinical evidence level
(https://civic.readthedocs.io/en/latest/model/evidence/level.html) as previously described5. In our study we only considered A and B levels of evidence, which represent variants that have
been FDA approved for treatment and are currently being evaluated in a late-stage clinical trial, respectively. A variant was determined to be ‘on-label’ when the cancer type matches the
cancer type for which the treatment was approved for or is being investigated for, and ‘off-label’ otherwise. Only actionable variants of the sensitive category were considered (that is,
tumours containing the variant are sensitive to a certain treatment). Sample-level actionable variants such as TMB high/low or MSI status were not evaluated, because of their tendency to
overshadow the other variants, especially in the off-label category. Furthermore, wild-type actionable variants were not considered in this analysis for the same reason. Variants related to
gene expression or methylation were not considered due to lack of available data. In addition, we found actionable variants derived from leukaemias to be very different from the solid
tumours in our dataset, which is why we excluded them for this analysis. For the analysis of proportion of samples bearing therapeutically actionable variants, we considered that the highest
evidence level was retained for each sample following the order A on/off-label to B on/off-label. To assess enrichment of actionable variants globally and at the A on-label level in
metastatic tumours, a Fisher’s exact test was performed pan-cancer-wide and per cancer type. An adjusted _P_ < 0.05 was deemed to be significant. Fold changes in frequency are only shown
for cancer types with a global significant difference. To determine which variants contribute the most to the observed significant frequency differences, individual actionable variants were
tested for enrichment in metastatic tumours using a Fisher’s exact test per cancer type and tier level. _P_ values were FDR adjusted per cancer type and _q_ < 0.05 was deemed to be
significant. In Extended Data Fig. 8, only actionable variants from cancer types with a global significant difference (see above) and that were found at a minimum frequency of 5% in either
primary or metastatic cohort and a minimum frequency difference of 5% between them were shown. However, the differences across all screened variants are available as part of Supplementary
Table 7. TEDS We aimed to pinpoint drivers that are potentially responsible for lack of response to certain cancer treatments in the metastatic cohort. Hence, we devised a test that
identifies driver alterations that are enriched in groups of patients treated with a particular treatment type compared with the untreated group of patients from the same cancer type (see
Extended Data Fig. 9a for illustration of the workflow). Treatments were grouped according to their mechanism of action so that multiple drugs with a shared mechanism of action were grouped
into the mechanistic treatment category (for example, cisplatin, oxaliplatin and carboplatin were grouped as platinum). We created 323 treatment and cancer-type groups by grouping patients
with treatment annotation according to their treatment record before the biopsy. One patient might be involved in multiple groups if they have received multiple lines of therapy or a
simultaneous combination of multiple drugs. Only 92 treatment and cancer-type groups with at least ten patients were further considered in the analysis. Hence, for each cancer type (or
subtype, in the case of breast and colorectal) and treatment group, we performed the following steps: * (1) We first performed a driver discovery analysis in treatment and cancer-type (or
subtype)-specific manner. We explored three types of somatic alterations: coding mutations, non-coding mutations and CN variants (see below for detailed description of each driver category).
Driver elements from each alteration category were selected for further analysis. * (2) For each driver alteration from (1), we compared the alteration frequency in the treated group to the
untreated group of the same cancer type. Each driver category (coding and non-coding mutations and CN variants) were evaluated independently. We performed a Fisher’s exact test to assess
the significance of the frequency differences. Similarly, we computed the odds ratio of the mutation frequencies for each driver alteration. The _P_ values were adjusted with a
multiple-testing correction using the Benjamini–Hochberg procedure (α = 0.05). An adjusted _P_ value of 0.05 was used for coding mutations and CN variants. An adjusted _P_ value of 0.1 was
used for non-coding variants due to the overall low mutation frequency of the elements included in this category, which hampered the identification of significant differences. * (3) We then
annotated each driver element with information about the exclusivity in the treatment group. We labelled drivers as treatment exclusive if the mutation frequency in the untreated group was
lower than 5% or we annotated as treatment enriched otherwise. In addition, we manually curated the identified drivers with literature references of their association with each treatment
category. * (4) Finally, the overlap of patients in multiple treatment groups (see above) in the same cancer type prompted us to prioritize the most significant treatment association for
each driver gene in a particular cancer type. In other words, for each driver gene that was deemed as significantly associated with multiple treatment groups in the same cancer type, we
selected the most significant treatment association, unless a driver-treatment annotation was clearly reported in the literature. The full catalogue of TEDs and their mutation frequencies
can be found in Supplementary Table 8. CODING MUTATION DRIVERS We used dNdScv (v0.0.1)60 with default parameters to identify cancer driver genes from coding mutations. A global _q_ < 0.1
was used as a threshold for significance. Mutation frequencies for each driver gene were extracted from the dNdScv output. We defined the mutation frequency as the number of samples bearing
non-synonymous mutations. NON-CODING MUTATION DRIVERS We used ActiveDriverWGS61 (v1.1.2, default parameters) to identify non-coding driver elements in five regulatory regions of the genome
including 3′ untranslated regions (UTRs), 5′ UTRs, long non-coding RNAs, proximal promoters and splice sites. For each element category, we extracted the genomic coordinates from Ensembl
v101. Each regulatory region was independently tested. To select for significant hits, we filtered on adjusted _P_ values (FDR < 0.1) and a minimum of three mutated samples. We defined
the mutation frequency as the number of mutated samples for each significantly mutated element in the treatment group. CN VARIANT DRIVERS We ran GISTIC2 (ref. 62) (v2.0.23) on each of the 92
treatment and cancer-type groups using the following settings: gistic2 -b <inputPath> -seg <inputSegmentation> -refgene hg19.UCSC.add_miR.140312.refgene.mat -genegistic 1 -gcm
extreme -maxseg 4000 -broad 1 -brlen 0.98 -conf 0.95 -rx 0 -cap 3 -saveseg 0 -armpeel 1 -smallmem 0 -res 0.01 -ta 0.1 -td 0.1 -savedata 0 -savegene 1 -qvt 0.1. The focal GISTIC peaks (_q_ ≤
0.1 and <1 Mb) were then annotated with functional elements using the coordinates from Ensembl v101. The frequency differences between treated and untreated cohorts on every gene was
assessed with Fisher’s exact test as described above. For this, we first calculated the focal amplification and deep depletion status of every gene within each sample. A gene was amplified
when the ploidy level of the gene was 2.5 ploidy levels higher than its genome-wide mean ploidy level (as measured by PURPLE), and deleted when the gene ploidy level was lower than 0.3 (that
is, deep deletion). We observed that the majority of the peaks contained multiple significant gene candidates (after multiple correction _q_ < 0.05) and therefore we retained the gene
most closely positioned to the peak summit, which is the most significantly enriched region across the treated samples. Next, we also found recurrent peaks across multiple treatment groups
per cancer type that are not, or less, present in the untreated control group because most of the Hartwig samples have received multiple treatment types. We therefore merged peaks with
overlapping ranges to produce a single peak per genomic region per cancer type. For each collapsed peak, we selected the treatment type showing the lowest _q_ value for the gene near the
peak summit. Deletion and amplification peaks were processed separately. GROUP-LEVEL AGGREGATION OF TREATMENT RESISTANCE-ASSOCIATED VARIANTS To estimate the contribution of TEDs to the total
number of drivers per sample in the metastatic cohort, we excluded any TED from the catalogue of driver mutations (see the above section ‘Driver alterations’) in a cancer-type-specific,
gene-specific and driver-type-specific manner. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA
AVAILABILITY Metastatic WGS data and metadata from the Hartwig Medical Foundation are freely available for academic use through standardized procedures. Request forms can be found at
https://www.hartwigmedicalfoundation.nl/en/data/data-acces-request/. Somatic variant calls, gene driver lists, CN profiles and other core data of the PCAWG cohort generated by the Hartwig
analytical pipeline are available for download at https://dcc.icgc.org/releases/PCAWG/Hartwig. Researchers will need to apply to the ICGC data access compliance office
(https://daco.icgc-argo.org) for the ICGC portion of the dataset. Similarly, users with authorized access can download the TCGA portion of the PCAWG dataset at
https://icgc.bionimbus.org/files/5310a3ac-0344-458a-88ce-d55445540120. Additional information on accessing the data, including raw read files, can be found at
https://docs.icgc.org/pcawg/data/. References and download links to the original independent datasets used in the analyses are included in each of the pertinent sections of the Methods and
Supplementary Notes, and a full list of all datasets used in the present study can be found in the data availability section of the Reporting Summary file. CODE AVAILABILITY The Hartwig
analytical processing pipeline is available (https://github.com/hartwigmedical/pipeline5) and implemented in Platinum (https://github.com/hartwigmedical/platinum). The source code to
reproduce the analysis of the manuscript is available in the following repository: https://github.com/UMCUGenetics/primary-met-wgs-comparison. REFERENCES * Lambert, A. W., Pattabiraman, D.
R. & Weinberg, R. A. Emerging biological principles of metastasis. _Cell_ 168, 670–691 (2017). CAS PubMed PubMed Central Google Scholar * Massagué, J. & Obenauf, A. C. Metastatic
colonization by circulating tumour cells. _Nature_ 529, 298–306 (2016). ADS PubMed PubMed Central Google Scholar * Welch, D. R. & Hurst, D. R. Defining the hallmarks of metastasis.
_Cancer Res._ 79, 3011–3027 (2019). CAS PubMed PubMed Central Google Scholar * ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. _Nature_
578, 82–93 (2020). ADS Google Scholar * Priestley, P. et al. Pan-cancer whole-genome analyses of metastatic solid tumours. _Nature_ 575, 210–216 (2019). ADS CAS PubMed PubMed Central
Google Scholar * Alexander, S. & Friedl, P. Cancer invasion and resistance: interconnected processes of disease progression and therapy failure. _Trends Mol. Med._ 18, 13–26 (2012).
PubMed Google Scholar * Turajlic, S. & Swanton, C. Metastasis as an evolutionary process. _Science_ 352, 169–175 (2016). ADS CAS PubMed Google Scholar * Massagué, J., Batlle, E.
& Gomis, R. R. Understanding the molecular mechanisms driving metastasis. _Mol. Oncol._ 11, 3–4 (2017). PubMed PubMed Central Google Scholar * Birkbak, N. J. & McGranahan, N.
Cancer genome evolutionary trajectories in metastasis. _Cancer Cell_ 37, 8–19 (2020). CAS PubMed Google Scholar * Weiss, F., Lauffenburger, D. & Friedl, P. Towards targeting of shared
mechanisms of cancer metastasis and therapy resistance. _Nat. Rev. Cancer_ 22, 157–173 (2022). CAS PubMed Google Scholar * Bertucci, F. et al. Genomic characterization of metastatic
breast cancers. _Nature_ 569, 560–564 (2019). ADS CAS PubMed Google Scholar * Robinson, D. R. et al. Integrative clinical genomics of metastatic cancer. _Nature_ 548, 297–303 (2017). ADS
CAS PubMed PubMed Central Google Scholar * Zehir, A. et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. _Nat. Med._ 23,
703–713 (2017). CAS PubMed PubMed Central Google Scholar * Cameron, D. L. et al. GRIDSS2: comprehensive characterisation of somatic structural variation using single breakend variants
and structural variant phasing. _Genome Biol._ 22, 202 (2021). CAS PubMed PubMed Central Google Scholar * Garcia-Prieto, C. A., Martínez-Jiménez, F., Valencia, A. & Porta-Pardo, E.
Detection of oncogenic and clinically actionable mutations in cancer genomes critically depends on variant calling tools. _Bioinformatics_ https://doi.org/10.1093/bioinformatics/btac306
(2022). * Nguyen, B. et al. Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. _Cell_ 185, 563–575.e11 (2022). CAS PubMed PubMed
Central Google Scholar * Roepman, P. et al. Clinical validation of whole genome sequencing for cancer diagnostics. _J. Mol. Diagn._ 23, 816–833 (2021). CAS PubMed Google Scholar *
Ben-David, U. & Amon, A. Context is everything: aneuploidy in cancer. _Nat. Rev. Genet._ 21, 44–62 (2020). CAS PubMed Google Scholar * Steiner, T. et al. Gain in chromosome 8q
correlates with early progression in hormonal treated prostate cancer. _Eur. Urol._ 41, 167–171 (2002). CAS PubMed Google Scholar * Taylor, A. M. et al. Genomic and functional approaches
to understanding cancer aneuploidy. _Cancer Cell_ 33, 676–689.e3 (2018). CAS PubMed PubMed Central Google Scholar * Steele, C. D. et al. Signatures of copy number alterations in human
cancer. _Nature_ 606, 984–991 (2022). ADS CAS PubMed PubMed Central Google Scholar * Gemble, S. et al. Genetic instability from a single S phase after whole-genome duplication. _Nature_
604, 146–151 (2022). * Donehower, L. A. et al. Integrated analysis of TP53 gene and pathway alterations in The Cancer Genome Atlas. _Cell Rep._ 28, 1370–1384.e5 (2019). CAS PubMed PubMed
Central Google Scholar * Kocakavuk, E. et al. Radiotherapy is associated with a deletion signature that contributes to poor outcomes in patients with cancer. _Nat. Genet._ 53, 1088–1096
(2021). CAS PubMed PubMed Central Google Scholar * Christensen, S. et al. 5-Fluorouracil treatment induces characteristic T>G mutations in human cancer. _Nat. Commun._ 10, 4571
(2019). ADS PubMed PubMed Central Google Scholar * Kucab, J. E. et al. A compendium of mutational signatures of environmental agents. _Cell_ 177, 821–836.e16 (2019). CAS PubMed PubMed
Central Google Scholar * Moore, L. et al. The mutational landscape of human somatic and germline cells. _Nature_ 597, 381–386 (2021). ADS CAS PubMed Google Scholar * Blokzijl, F. et
al. Tissue-specific mutation accumulation in human adult stem cells during life. _Nature_ 538, 260–264 (2016). ADS CAS PubMed PubMed Central Google Scholar * Abascal, F. et al. Somatic
mutation landscapes at single-molecule resolution. _Nature_ 593, 405–410 (2021). ADS CAS PubMed Google Scholar * Alexandrov, L. B. et al. Clock-like mutational processes in human somatic
cells. _Nat. Genet._ 47, 1402–1407 (2015). CAS PubMed PubMed Central Google Scholar * Klein, C. A. Parallel progression of primary tumours and metastases. _Nat. Rev. Cancer_ 9, 302–312
(2009). CAS PubMed Google Scholar * Berges, R. R. et al. Implication of cell kinetic changes during the progression of human prostatic cancer. _Clin. Cancer Res._ 1, 473–480 (1995). ADS
CAS PubMed PubMed Central Google Scholar * Nguyen, L., W M Martens, J., Van Hoeck, A. & Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. _Nat. Commun._ 11,
5584 (2020). ADS CAS PubMed PubMed Central Google Scholar * Menghi, F. et al. The tandem duplicator phenotype is a prevalent genome-wide cancer configuration driven by distinct gene
mutations. _Cancer Cell_ 34, 197–210.e5 (2018). CAS PubMed PubMed Central Google Scholar * Degasperi, A. et al. A practical framework and online tool for mutational signature analyses
show inter-tissue variation and driver dependencies. _Nat. Cancer_ 1, 249–263 (2020). CAS PubMed PubMed Central Google Scholar * Behjati, S. et al. Mutational signatures of ionizing
radiation in second malignancies. _Nat. Commun._ 7, 12605 (2016). ADS CAS PubMed PubMed Central Google Scholar * Jeselsohn, R., Buchwalter, G., De Angelis, C., Brown, M. & Schiff,
R. ESR1 mutations—a mechanism for acquired endocrine resistance in breast cancer. _Nat. Rev. Clin. Oncol._ 12, 573–583 (2015). CAS PubMed PubMed Central Google Scholar * Watson, P. A.,
Arora, V. K. & Sawyers, C. L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. _Nat. Rev. Cancer_ 15, 701–711 (2015). CAS PubMed PubMed Central
Google Scholar * Yun, C.-H. et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. _Proc. Natl Acad. Sci. USA_ 105, 2070–2075 (2008). ADS CAS
PubMed PubMed Central Google Scholar * Nathanson, D. A. et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. _Science_ 343, 72–76 (2014).
ADS CAS PubMed Google Scholar * Watson, R. G. et al. Amplification of thymidylate synthetase in metastatic colorectal cancer patients pretreated with 5-fluorouracil-based chemotherapy.
_Eur. J. Cancer_ 46, 3358–3364 (2010). CAS PubMed PubMed Central Google Scholar * Yang, L. et al. lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs.
_Nature_ 500, 598–602 (2013). ADS CAS PubMed PubMed Central Google Scholar * Xiao, Y., Lin, F.-T. & Lin, W.-C. ACTL6A promotes repair of cisplatin-induced DNA damage, a new
mechanism of platinum resistance in cancer. _Proc. Natl Acad. Sci. USA_ 118, e2015808118 (2021). CAS PubMed PubMed Central Google Scholar * Formisano, L. et al. Aberrant FGFR signaling
mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. _Nat. Commun._ 10, 1373 (2019). ADS PubMed PubMed Central Google Scholar * Jamal-Hanjani, M. et al. Tracking the evolution
of non-small-cell lung cancer. _N. Engl. J. Med._ 376, 2109–2121 (2017). CAS PubMed Google Scholar * GLASS Consortium. Glioma through the looking GLASS: molecular evolution of diffuse
gliomas and the Glioma Longitudinal Analysis Consortium. _Neuro Oncol._ 20, 873–884 (2018). Google Scholar * Kim, N. et al. Single-cell RNA sequencing demonstrates the molecular and
cellular reprogramming of metastatic lung adenocarcinoma. _Nat. Commun._ 11, 2285 (2020). ADS CAS PubMed PubMed Central Google Scholar * Moldoveanu, D. et al. Spatially mapping the
immune landscape of melanoma using imaging mass cytometry. _Sci. Immunol._ 7, eabi5072 (2022). CAS PubMed Google Scholar * Martínez-Jiménez, F. et al. Genetic immune escape landscape in
primary and metastatic cancer. _Nat. Genet._ https://doi.org/10.1038/s41588-023-01367-1 (2023). * Nguyen, L., Van Hoeck, A. & Cuppen, E. Machine learning-based tissue of origin
classification for cancer of unknown primary diagnostics using genome-wide mutation features. _Nat. Commun._ 13, 4013 (2022). ADS CAS PubMed PubMed Central Google Scholar * Alexandrov,
L. B. et al. The repertoire of mutational signatures in human cancer. _Nature_ 578, 94–101 (2020). ADS CAS PubMed PubMed Central Google Scholar * Signal. _Signal_
https://signal.mutationalsignatures.com/explore/study/6?mutationType=1 (accessed 1 February 2023). * Forbes, S. A. et al. COSMIC: somatic cancer genetics at high-resolution. _Nucleic Acids
Res._ 45, D777–D783 (2017). CAS PubMed Google Scholar * Gerstung, M. et al. The evolutionary history of 2,658 cancers. _Nature_ 578, 122–128 (2020). ADS CAS PubMed PubMed Central
Google Scholar * Shale, C. et al. Unscrambling cancer genomes via integrated analysis of structural variation and copy number. _Cell Genomics_ 2, 100112 (2022). CAS PubMed PubMed Central
Google Scholar * Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of _Drosophila melanogaster_
strain w1118; iso-2; iso-3. _Fly_ 6, 80–92 (2012). CAS PubMed PubMed Central Google Scholar * Chakravarty, D. et al. OncoKB: a precision oncology knowledge base. _JCO Precis. Oncol._
2017, PO.17.00011 (2017). PubMed Google Scholar * Griffith, M. et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. _Nat.
Genet._ 49, 170–174 (2017). CAS PubMed PubMed Central Google Scholar * Tamborero, D. et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor
alterations. _Genome Med._ 10, 25 (2018). PubMed PubMed Central Google Scholar * Martincorena, I. et al. Universal patterns of selection in cancer and somatic tissues. _Cell_ 171,
1029–1041.e21 (2017). CAS PubMed PubMed Central Google Scholar * Zhu, H. et al. Candidate cancer driver mutations in distal regulatory elements and long-range chromatin interaction
networks. _Mol. Cell_ 77, 1307–1321.e10 (2020). CAS PubMed Google Scholar * Mermel, C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic
copy-number alteration in human cancers. _Genome Biol._ 12, R41 (2011). PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This publication and the underlying
study have been made possible partly on the basis of the data that Hartwig Medical Foundation and the Center of Personalized Cancer Treatment (CPCT) have made available to the study. We
thank R. Janssen for the technical assistance in the collection and processing of the PCAWG raw sequencing data using the Hartwig tumour analytical pipeline; USEQ from UMC Utrecht for
providing high-performance computing power; J. Mateo for his valuable scientific input; L. Stein and L. Xiang for their assistance in the publication of the reprocessed ICGC part of the
PCAWG dataset; R. Grossman, C. Meyer and T. Simmons for their assistance in the publication of the reprocessed TCGA part of the PCAWG; D. Quigley for providing the somatic mutation calls and
age at biopsy of the patients with prostate cancer from their study; S. Turajlic and H. Pallikonda for their responsiveness concerning the TRACERx renal dataset; and P. Wolfe and other
staff of the Hartwig Medical Foundation team for aligning the processing of PCAWG and Hartwig datasets. AUTHOR INFORMATION Author notes * These authors contributed equally: Ali Movasati,
Sascha Remy Brunner, Luan Nguyen AUTHORS AND AFFILIATIONS * Center for Molecular Medicine, Oncode Institute, University Medical Center Utrecht, Utrecht, The Netherlands Francisco
Martínez-Jiménez, Ali Movasati, Sascha Remy Brunner, Luan Nguyen, Edwin Cuppen & Arne Van Hoeck * Vall d’Hebron Institute of Oncology (VHIO), Barcelona, Spain Francisco Martínez-Jiménez
* Hartwig Medical Foundation, Amsterdam, The Netherlands Francisco Martínez-Jiménez & Edwin Cuppen * Hartwig Medical Foundation Australia, Sydney, New South Wales, Australia Luan Nguyen
& Peter Priestley Authors * Francisco Martínez-Jiménez View author publications You can also search for this author inPubMed Google Scholar * Ali Movasati View author publications You
can also search for this author inPubMed Google Scholar * Sascha Remy Brunner View author publications You can also search for this author inPubMed Google Scholar * Luan Nguyen View author
publications You can also search for this author inPubMed Google Scholar * Peter Priestley View author publications You can also search for this author inPubMed Google Scholar * Edwin Cuppen
View author publications You can also search for this author inPubMed Google Scholar * Arne Van Hoeck View author publications You can also search for this author inPubMed Google Scholar
CONTRIBUTIONS F.M.-J., A.V.H. and E.C. conceptualized the study. F.M.-J., A.M., S.R.B., L.N., P.P. and A.V.H. came up with the methodology. F.M.-J., A.M., S.R.B., L.N., P.P. and A.V.H.
provided software. F.M.-J., A.M., S.R.B., L.N., P.P. and A.V.H. validated results. F.M.-J., A.M., S.R.B., L.N., P.P., A.V.H. and E.C. conducted formal analysis. F.M.-J., A.M., S.R.B., L.N.,
P.P. and A.V.H. performed the investigations and provided resources. F.M.-J., A.M., S.R.B., L.N., P.P. and A.V.H. curated the data. F.M.-J. and A.V.H. wrote the original draft of the
manuscript. F.M.-J., A.M., S.R.B., L.N., P.P., A.V.H. and E.C. reviewed and edited the manuscript. F.M.-J., A.M., S.R.B., L.N., A.V.H. and E.C. performed visualization. F.M.-J., A.V.H. and
E.C. supervised the project. A.V.H. and E.C. provided project administration. E.C. acquired funding. CORRESPONDING AUTHOR Correspondence to Edwin Cuppen. ETHICS DECLARATIONS COMPETING
INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature_ thanks Peter Van Loo and the other, anonymous, reviewer(s) for their contribution to the
peer review of this work. Peer reviewer reports are available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps
and institutional affiliations. EXTENDED DATA FIGURES AND TABLES EXTENDED DATA FIG. 1 COHORT OVERVIEW AND GLOBAL GENOMIC FEATURES. A) Workflow of the unified processing pipeline used in this
study for Hartwig (left) and PCAWG (right) WGS samples. First, PCAWG tumor and matched normal raw sequencing files were gathered and re-processed using the Hartwig tumor analytical
pipeline. Next, the output of tumor samples that were correctly processed by the pipeline were further subjected to a strict quality control filtering. As a result, a total of 7,108 samples
from 71 cancer types compose the harmonized dataset. 5,365 patient tumor samples from 23 cancer types with sufficient representation in both primary and metastatic datasets were selected for
this study. B) Tumor clonality according to the metastatic biopsy location in kidney renal clear cell carcinoma, lung adenocarcinoma, prostate carcinoma and skin melanoma. N, number of
samples in the group. p, two-sided Mann-Whitney p-value. Box-plots: center line, median; box limits, first and third quartiles; whiskers, lowest/highest data points at first quartile
minus/plus 1.5× IQR. C) Left, similar to Fig. 1d only including non-WGD tumors. Right, similar to Fig. 1e for non-WGD tumors. D) Equivalent to c), but limited to WGD tumors. ‘*’, two-sided
Mann-Whitney adjusted p-value < 0.01 for continuous variables and two-sided Fisher’s exact test adjusted p-value < 0.01 for _TP53_. EXTENDED DATA FIG. 2 MUTATION BURDEN AND MUTATIONAL
SIGNATURES. A) double-base substitutions (DBSs, top) and indels (IDs, bottom) mutational spectra of metastatic and primary tumors. Patients are ordered according to their TMB burden. B) Moon
plot representing the DBS burden differences attributed to each mutational signature in metastatic (left) and primary (right) tumors. Edge thickness and colors represent significant
differences (Mann-Whitney adjusted p-value<0.05, ±1.4x fold change) and the direction of the enrichment, respectively. The size of circles are proportionate to the mutation burden
difference. Right bars, number of metastatic cancer types with a mutational signature significant enrichment. Top stacked bars represent the cumulative signature exposure difference. Thicker
bar edge lines represent significance. Bars are coloured according to the annotated etiology. Only mutational signatures with known etiology or with at least one cancer type with
significant metastatic enrichment are included. C) analogous representation for IDs. D) Volcano plot representing the mutational signature hypermutation (>10,000 mutations for SBS,
>500 for DBS, and >1000 for ID) prevalence comparison between primary and metastatic tumor patients. Y-axis, log10(two-sided Mann-Whitney adjusted p-value). X-axis, effect size as
Cramer’s V. Each dot represents a mutational signature in a cancer type. Dots are coloured according to the mutation type. Diff., difference. Muts. mutations. Sig., mutational signature.
Mut. mutational. Susp., suspected. Def., deficiency. EXTENDED DATA FIG. 3 MUTATIONAL SIGNATURE RELATIVE CONTRIBUTION COMPARISON. A) From top to bottom, moon plot representing the SBS, DBS
and ID relative contribution differences attributed to each mutational signature. The size of circles are proportional to the relative mutation burden difference. Top stacked bars represent
the relative signature exposure difference. Thicker bar edge lines represent significance (two-sided Mann-Whitney adjusted p-value < 0.05 and ≥1% difference in relative contribution).
Bars are coloured according to the annotated etiology. Right bars, number of metastatic cancer types with a mutational signature significant enrichment. Only mutational signatures with known
etiology or with at least one cancer type with significant enrichment are included. Diff., difference. Muts. mutations. Sig., mutational signature. Mut. mutational. Susp., suspected. Def.,
deficiency. EXTENDED DATA FIG. 4 AGE-CORRECTED SBS1 MUTATION BURDEN IN PRIMARY AND METASTATIC TUMORS. A) Linear regression of the SBS1 mutation burden (y-axis) and patient’s age at biopsy
(x-axis) in primary and metastatic cancer across the 23 cancer types. The median trendline and 99% confidence intervals of the linear regression are represented as a solid line and the
adjacent shaded area, respectively. The mean fold change, mean SBS1 increase per year and one sided Mann-Whitney p-value are only displayed in cancer types with an age-independent
significantly different primary and metastatic distribution. Red labels, significant increase in metastatic tumors. Blue, control cancer types. Rmet and Rprim, Pearson correlation
coefficient of the metastatic and primary linear regressions, respectively. B) Analogous representation for independent linear regressions for breast cancer subtypes. C) Relative to a) for
ploidy corrected SBS1 in the tumor types of interest. D) Relative to a) for ploidy corrected SBS5/40 counts in the tumor types of interest. E) Depiction illustrating the potential effect of
an increased cell division rate in metastatic tumors compared to primary and its expected impact on the SBS1 variant allele frequency (VAF) distribution. Partially created BioRender.com. F)
Comparison of global SBS1 clonality ratios between primary and metastatic in breast, prostate, kidney renal clear cell, thyroid, colorectal and ovarian serous carcinomas. Boxplots are
defined as in Fig. 1. P, two-sided Mann-Whitney p-value. N, number of samples. G) Spearman correlation analysis of the mean SBS1 year burden of primary tumors (y-axis) and the mean
metastatic SBS1 fold change (x-axis) across the 15 cancer types with linear association between age and SBS1 accumulation. Vertical error bars represent the 25th and 75th percentile,
respectively. Horizontal error bars represent the standard deviation of the mean fold change (metastatic divided by primary) of the SBS1 yearly mutation burden. The median trendline and 99%
confidence intervals of the linear regression are represented as a solid line and the adjacent shaded area, respectively. Cancer types with a significantly different SBS1 mutation rate are
marked by thicker marker borders and with red labels. Blue labels represent the control cancer types. H) Similar but using SBS1 year mutation rate from ref. 30. To derive vertical and
horizontal error bars in panels g) and h) all tumor samples from the primary and metastatic cohorts from panel a) (see Methods for inclusion criteria) were included in the analysis. The
number of included samples per cancer type and cohort are available in Supplementary Table 4. Muts, mutations. EXTENDED DATA FIG. 5 STRUCTURAL VARIANT BURDEN. A) Top rectangles represent the
four genomic instability features defined in Fig. 1e. A red background represents significant enrichment in the metastatic cohort (two-sided Mann-Whitney adjusted p-value <0.01).
S-plots, cumulative distribution function plot (samples ranked independently for each SV type) of tumor mutation burden for each cancer type for (from top to the bottom) the aggregated
structural variant (SV) burden, small deletions (<10kb), large deletions (>=10kb), small duplications (<10kb), large duplications (>=10kb), complex events (<20 breakpoints),
complex events (>=20 breakpoints) and LINEs insertions. Horizontal lines represent median values. Backgrounds are coloured according to the relative enrichment, defined as: log10(median
SV type burden in metastatic tumors + 1) − log10(median SV type burden in primary tumors + 1). Fold change labels and coloured backgrounds are displayed when Mann-Whitney comparison renders
a significant q-value < 0.05. Fold change labels are displayed with ‘>’ when the SV burden for primary tumors is 0 (see Methods for more details). For each cancer type, bottom bar
plots represent the relative fraction of each SV type in the metastatic (left) and primary (right) datasets. B) SV length frequency distribution of deletions (left panel) and duplications
(middle panel). Right panel shows the frequency distribution of the number of linked breakpoints for complex SVs. Dashed vertical lines represent the chosen threshold to separate between
short and large deletions, duplications and complex SVs, respectively. EXTENDED DATA FIG. 6 STRUCTURAL VARIANT BURDEN ASSOCIATED GENOMIC FEATURES. A) Volcano plot representing the cancer
type-specific regression coefficients (x-axis) and significance (y-axis, measured by the linear regression model coefficient p-value) of clinical and genomic features against the number of
small deletions. Each dot represents one feature in one cancer type. Labels are coloured according to the feature category. Dots are coloured by the frequency enrichment in metastatic
(purple) or primary (orange) patients. Analogous panels are displayed for B) large deletions, C) short duplications, D) large duplications, E) short complex SVs, F) large complex SVs and G)
LINEs. H) Lollipop plots representing the regression coefficients (left, relative to panel b. x-axis) and metastatic enrichment (right, relative to dots colour from panel b.) of features
associated with small deletions. Only significant features (LM>0.0, LM coefficient p-value < 0.01 and with independent significance in primary or metastatic tumors) enriched in
metastatic tumor patients (enrichment > 0.0) are displayed. I), J) and K) are identical but referring to large deletions, small duplications and large duplications, respectively. LM,
linear model. Coef, coefficient. EXTENDED DATA FIG. 7 DRIVER LANDSCAPE AND DRIVERS PER PATIENT. A) Cancer type-specific distribution of number of driver alterations, amplifications,
deletions and mutations per patient in primary (top) and metastatic (bottom). Black dots represent the mean values. Labels display mean differences (metastatic - primary) in cancer types
with a significant difference. “*”, two-sided Mann-Whitney adjusted p-value < 0.01. B) Volcano plots representing the cancer type-specific enrichment (x-axis) and significance (y-axis,
FDR adjusted two-tailed Fisher’s exact test p-value) of driver genes between primary and metastatic cohorts. From left to right, amplification drivers, biallelically deleted drivers and
mutated driver genes. BRCA, Breast carcinoma. KIRC, kidney renal clear cell carcinoma. OV, Ovarian serous adenocarcinoma. PRAD, Prostate carcinoma. SKCM, Skin melanoma. THCA, Thyroid
carcinoma. LIHC, Hepatocellular carcinoma. PANET, pancreatic neuroendocrine. EXTENDED DATA FIG. 8 THERAPEUTIC ACTIONABILITY OF VARIANTS. A) Cancer type-specific fraction of primary (top) and
metastatic (bottom) patients with reported therapeutically actionable variants. For each patient the variant with the greatest level of evidence was considered. Bars are coloured according
to the variant actionability tiers. Fold change (i.e., metastatic divided by primary fraction) labels are displayed in cancer types with a significant proportional increase (two-sided
Fisher’s exact test adjusted p-value < 0.05). Purple edgelines highlight significant increase in metastatic A-on label fraction patients. B) Primary (left) and metastatic (right)
alteration frequency of actionable variants with a high discrepancy (>5% frequency difference) from cancer types with a global significant increase of actionable variants in metastatic
tumor patients from panel a). “*”, a two-sided Fisher’s exact test adjusted p-value < 0.05. Text boxes include the associated treatments for alterations with a significant mutation
frequency increase in metastatic tumor patients. EXTENDED DATA FIG. 9 TREATMENT ENRICHED DRIVERS. A) Visual depiction of the analytical framework to identify treatment enriched drivers
(TEDs). Example, identification of TEDs in the 354 breast carcinoma patients treated with aromatase inhibitors. (1), identification of cancer driver genes from coding mutations (green),
non-coding mutations (soft green), copy number amplifications (red) and deletions (blue). (2), for each driver gene, comparison of the alteration frequency in treated and untreated patients.
(3) and (4), annotation of TEDs with type of enrichment and orthogonal evidence B) Side by side alteration frequency comparison between treated (right bar) and untreated (left bar) patients
for all treatment-exclusive and C) treatment-enriched TEDs. D) Distribution of mutations along the _AR_ protein sequence in prostate cancer patients treated with androgen deprivation (top)
and untreated (bottom). Pfam domains are represented as rectangles. Mutations are coloured according to the consequence type. E) Distribution of focal copy number gains in chromosome X in
prostate untreated patients (bottom) and treated with androgen deprivation (top). _AR_ coding region and the promoter region are highlighted. F) Distribution of mutations along the _ESR1_
protein sequence in breast carcinoma patients treated with aromatase inhibitors (top) and untreated (bottom). G) Distribution of mutations along the _EGFR_ protein in lung adenocarcinoma
patients treated with EGFR inhibitors (top) and untreated (bottom). Pfam domains are represented as rectangles. H) Distribution of focal copy number gains in chromosome 7 in lung
adenocarcinoma untreated patients (bottom) and treated with anti-EGFR (top). _EGFR_, _MET_ and _CDK6_ genomic locations are highlighted. I) Distribution of focal copy number gains in
chromosome chr18p:1Mb-8Mb in breast carcinoma untreated patients (bottom) and treated with pyrimidine antagonists (top). TYMS genomic location is highlighted. J) Similar to f) but
representing ultra-focal (shorter than 3Mbs) MYC and PRNCR1 amplifications in chromosome 8q. In all copy number gain plots each bin represents 100Kbs. Mb, megabase. Kb, kilobase. EXTENDED
DATA FIG. 10 PAN-CANCER DIFFERENCES BETWEEN PRIMARY AND METASTATIC TUMORS. A) Stacked plot representing the qualitative differences of the eight studied genomic features across the 23 cancer
types included in this study. Cancer types are sorted in ascending order according to the cumulative number of diverging genomic features between primary and metastatic tumors. Each
horizontal track represents a genomic feature. The presence (and height) of each feature for a specific cancer type correlates with the magnitude of the observed differences. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY INFORMATION This file contains Supplementary Notes 1-4 REPORTING SUMMARY PEER REVIEW FILE SUPPLEMENTARY DATA 1 Mutation profiles of de novo signatures that were
extracted with non-negative matrix factorization. SUPPLEMENTARY TABLE 1 Primary and metastatic cohorts metadata. SUPPLEMENTARY TABLE 2 Karyotype and genomic instability measurements.
SUPPLEMENTARY TABLE 3 Mutational signatures absolute and relative contribution. SUPPLEMENTARY TABLE 4 SBS1 mutation rate. SUPPLEMENTARY TABLE 5 Structural variants. SUPPLEMENTARY TABLE 6
Driver alterations. SUPPLEMENTARY TABLE 7 Therapeutic actionability of variants. SUPPLEMENTARY TABLE 8 Treatment associated drivers (TEDs). RIGHTS AND PERMISSIONS OPEN ACCESS This article is
licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in
this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative
Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a
copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Martínez-Jiménez, F., Movasati, A., Brunner, S.R. _et
al._ Pan-cancer whole-genome comparison of primary and metastatic solid tumours. _Nature_ 618, 333–341 (2023). https://doi.org/10.1038/s41586-023-06054-z Download citation * Received: 28
June 2022 * Accepted: 05 April 2023 * Published: 10 May 2023 * Issue Date: 08 June 2023 * DOI: https://doi.org/10.1038/s41586-023-06054-z SHARE THIS ARTICLE Anyone you share the following
link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature
SharedIt content-sharing initiative