Dynamics of heavy chain junctional length biases in antibody repertoires
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Antibody variable domain sequence diversity is generated by recombination of germline segments. The third complementarity-determining region of the heavy chain (CDR H3) is the
region of highest sequence diversity and is formed by the joining of heavy chain VH, DH and JH germline segments combined with random nucleotide trimming and additions between these
segments. We show that CDR H3 and junctional segment length distributions are biased in human antibody repertoires as a function of VH, VL and JH germline segment utilization. Most length
biases are apparent in the naive and antigen experienced B cell compartments but not in nonproductive recombination products, indicating B cell selection as a major driver of these biases.
Our findings reveal biases in the antibody CDR H3 diversity landscape shaped by VH, VL, and JH germline segment use during naive and antigen-experienced repertoire selection. SIMILAR CONTENT
BEING VIEWED BY OTHERS VHH CDR-H3 CONFORMATION IS DETERMINED BY VH GERMLINE USAGE Article Open access 19 August 2023 NEW INSIGHTS INTO ANTIBODY STRUCTURE WITH IMPLICATIONS FOR SPECIFICITY,
VARIABLE REGION RESTRICTION AND ISOTYPE CHOICE Article 20 March 2025 UNVEILING INVERTED D GENES AND D-D FUSIONS IN HUMAN ANTIBODY REPERTOIRES UNLOCKS NOVEL ANTIBODY DIVERSITY Article Open
access 28 January 2025 INTRODUCTION The diversity of sequences in the variable regions of immunoglobulins is the basis for the ability of these molecules to bind a virtually unlimited number
of antigenic structures. Sequence diversity in the primary repertoire is created by recombination of germline segments for both the heavy and light chains, which results in the formation of
full-length immunoglobulin variable region exons1. The light chain variable region is created by the joining of VL and JL germline segments while the VH region is created by recombination
of VH, DH, and JH germline segments. The process of recombination starts with the heavy chain in progenitor B cells, initiated by D/JH recombination followed by VH/DJH recombination2,3.
Light chain recombination occurs in pre-B cells after successful completion of the heavy chain recombination. Germline segments in both chains are also trimmed and extended by a variable
number of nucleotides by exonucleolytic nibbling of germline segments and random nucleotide incorporation in the N-regions flanking the DH germline segment mediated by terminal
deoxynucleotidyl transferase and germline segment palindromic duplications3. Self-reactive B cell clones with full-length, in-frame variable regions have VL sequences replaced by receptor
editing or are removed from the repertoire by apoptosis4,5. Cells passing this self-reactivity checkpoint form the naive B cell repertoire5. The third complementarity-determining region
(CDR) of the heavy chain (CDR H3) is the region of highest overall sequence and length diversity in antibody repertoires1. CDR H3 length approximates a Gaussian distribution6 and the average
CDR H3 length varies as a function of species, age, isotype, B cell development stage and disease state6,7,8,9,10,11,12,13. CDR H3 amino acid composition is also biased in a CDR H3
length-dependent manner, associated with differential incorporation of DH and JH germline segment sequences of different lengths and sequence composition into CDR H3 of different lengths6.
Beyond the germline segment biases, CDR H3 sequence biases that reflect underlying selective biases in B cell maturation have also been described. In particular, a bias towards shorter
average CDR H3 lengths is observed in mature relative to immature B cells and in isotype-switched memory B cells relative to naive to B cells9,10,14. This is accompanied by a reduction of
positively charged residue content and hydrophobicity within CDR H3 associated with negative selection of self-reactive clones in the repertoire9,11,15,16. The analyses of CDR H3 diversity
and biases in health and disease have been mostly performed independently of the V regions contributed by VH and VL germline segments6,7,8,9,10,11,17,18,19,20. Except for sequences that are
directly incorporated into CDR H3, the impact of V germline segments on CDR H3 properties has neither been addressed nor expected. Analysis of the impact of the VL on CDR H3 has been limited
to properties of the third CDR of the light chain, which is closely associated with CDR H3, without any evidence of biases14. Finally, analysis of the impact of JH germline segments on CDR
H3 biases has been confined to the expected effects of differential JH germline segment length and sequence composition6. A recent analysis of a large dataset of isotype-switched human
antibody sequences with paired chain information revealed an unexpected preferential pairing of _IGHV3-7_ (VH3-7) and Vκ2-30 germline segments21. This was determined upon further analysis to
be linked with short CDR H3 length biases associated with both germline segments. This unexpected finding prompted us to undertake a high-dimensional analysis of CDR H3 sequences from
several human antibody datasets to investigate the extent to which CDR H3 length might be biased by germline segment use in human immunoglobulin repertoires. Our results show several biases
in CDR H3 and junctional length distributions associated with VH, VL, and JH germline segment utilization that shape naive and antigen-experienced antibody repertoires in unexpected and
unpredictable patterns. RESULTS SEQUENCE DATASETS We analyzed sequences from four previously described B cell repertoire deep sequencing datasets including three donors each and a fifth
dataset with eight donors (used here as an independent test dataset in most analyses to avoid over-representation of donors from a single source in combined data), referred to here as the
TX, WA, CA, MA, and SRI datasets20,21,22,23,24,25. These represent the largest publicly available datasets with VH/VL pairing (TX and CA) or VH-only information (MA, WA, SRI). The datasets
were previously sequenced and bioinformatically parsed using a diversity of methods, minimizing the impact of methodological biases. A summary of the datasets including the number of donors,
CD27 marker and isotype information has been summarized in Table 1 (see Supplementary Table 1 for details). Data was normalized by analyzing each donor individually and averaging within or
among datasets to avoid over-representation of sequences from larger datasets. No antigen-specific selection of B cells was performed for any of the datasets, although the CA and MA datasets
include both pre- and post-vaccination samples21,22. For simplicity we refer to the TX CD27pos IgG/IgA, CA IgG, MA IgG/IgA, WA CD27pos, and SRI IgG subsets as TX AE, CA AE, MA AE, WA AE,
and SRI AE, respectively (with “antigen-experienced”, or “AE”, encompassing all five), the TX CD27pos IgM sequences as “AE IgM” and the TX and WA CD27neg subsets as TX naive and WA naive
respectively (with “naive” encompassing both, Supplementary Table 1). SRI IgM sequences with no amino acid somatic mutations between Cys-23 and Cys-104 (International ImMunoGeneTics, IMGT®,
numbering system26 used throughout) including most of the region covered by reads are referred as SRI naive. We aimed at identifying properties shared among donors not influenced by clonal
expansion. To minimize the impact of clonal expansion, all datasets except nonproductive sequences were processed to retain a single sequence from each lineage, or clonotype, by clonotype
clustering, according to germline segments as well as CDR H3 length and sequence similarity (see ‘Clonotype clustering’ under Methods and Supplementary Data 1, 2, and 3 for unique sequence
counts after clustering). Overall distribution of CDR H3 lengths was not noticeably affected by removal of redundant sequences in most datasets except for the WA AE and MA AE compartments,
which had subtle shifts (Supplementary Fig. 1a-f). The overall AE CDR H3 length distributions are similar among datasets except SRI, allowing pooling data of AE B cell subsets from different
datasets (Supplementary Fig. 1g). However, the relative CDR H3 length distributions of the WA and TX naive B cell subsets differed by an average of 0.9 residues (Supplementary Fig. 1h) and
were analyzed separately. Germline segment-specific analyses were performed with germline segments with at least 80 unique clonotypes in a donor, which, in aggregate, include 94–99% of the
unique clonotype sequences in the CA, TX, MA and SRI datasets (Supplementary Data 1, 2, and 3). Germline segment-specific analyses in the WA dataset were performed with 16 VH germline
segments that had fewer than 10% ambiguous calls in the naive subset and germline segments prevalent in other datasets, totaling about one third of the entire dataset (Supplementary Data 4).
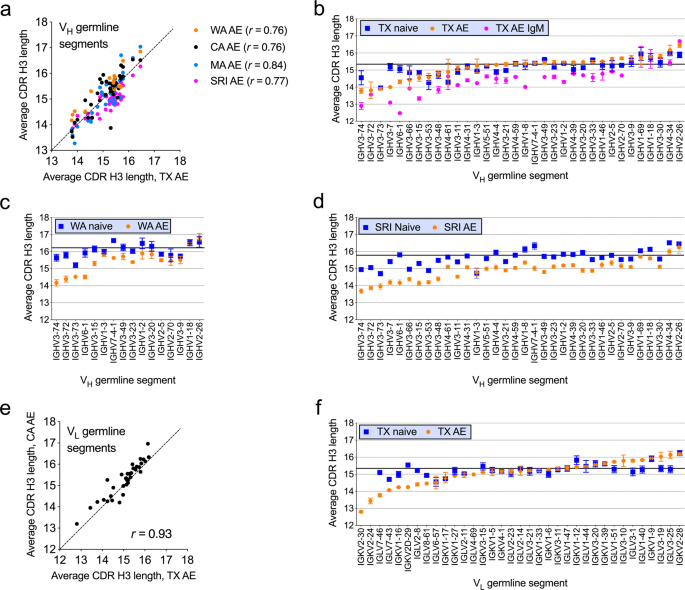
AVERAGE CDR H3 LENGTH VARIES WITH VH AND VL GERMLINE SEGMENT USE We analyzed average CDR H3 length by VH or VL germline segment use. Average CDR H3 length in the AE subset varied by up to 3
amino acid residues as a function of VH germline segment use and correlated well for different datasets when compared to the TX dataset (Spearman’s _r_ = 0.72–0.84) (Fig. 1a). Average CDR
H3 length also varied as a function of VL germline segment use by up to four amino acid residues in the AE compartment and correlated well between the CA and TX datasets (Spearman’s _r_ =
0.93) (Fig. 1e). The naive compartment showed a more limited spread in average CDR H3 lengths relative to the AE compartment (Fig. 1b, c, d and f, blue squares). More pronounced reductions
in average CDR H3 length in the AE relative to naive compartments were associated with a subset of VH and VL germline segments (Fig. 1b, c, d and f). The TX AE IgM subset showed similar
trends as the TX AE subset except that average CDR H3 length was decreased in association with most VH germline segments relative to the naive compartment (Fig. 1b, pink dots). CDR H3 LENGTH
DISTRIBUTION VARIES WITH VH GERMLINE SEGMENT USE We next determined whether CDR H3 length distribution varies with germline segment use. Overall and germline segment-specific CDR H3 length
distributions were performed individual for each donor and the frequency of each CDR H3 length averaged within or across the TX, CA, MA, and WA datasets (11 donors), with the eight donors
from the SRI dataset analyzed separately. Overall CDR H3 length distribution of the respective B cell compartment, which is influenced by germline segment frequency in the datasets, was used
as a relative standard to which germline segment-specific CDR H3 length distributions were compared. This was done to facilitate comparison of biases between germline segments and also
because useful objective reference distributions are not available to determine bias types in naive compartment sequences. Therefore, most biases described here, including all in the naive
compartment, are relative to the entire set of clonotypes in each B cell compartment. Statistical analysis of biases was performed in the AE compartment by a two-tailed paired (by donor)
_t_-test of length frequencies with a sliding window of two consecutive CDR H3 lengths to minimize the impact of local fluctuations. Observed length distribution biases included overall
shifts in average CDR H3 length for sequences with different VH germline segments and also obvious and subtle deviations from the overall CDR H3 distribution in discrete ranges of the length
spectrum (Fig. 2, top row, Supplementary Fig. 2a). To further discern the CDR H3 length biases quantitatively, we performed a principal component (PC) analysis of the length distributions
(lengths 5–26) associated with different VH germline segments. Results from the PC analysis were visualized by projecting each germline segment onto the most important trends to obtain the
so-called PC scores, aided by a visual analysis of the corresponding distributions (Fig. 3a). PC1 and PC2 corresponded to apparent skewness and kurtosis of the distributions, respectively.
Using the PC analysis results in conjunction with visual inspection of VH germline segment-associated CDR H3 distributions in the AE compartment, germline segments were categorized by bias
type as “Short”, “Neutral” and “Long” (Figs. 2 and 3a, Supplementary Fig. 2). Those germlines that had similar length distributions as the overall distribution were called “Neutral” (located
around the center of the PC plot); while those with shifts towards longer or shorter lengths as “Long” (right-skewed, with low values of PC1) and “Short” (left-skewed, with high values of
PC1) respectively. Within each of these classes, some germlines also showed varying degrees of kurtosis relative to the overall distribution (extreme values in PC2). Differences between the
distributions of members of different groups can be subtle, both visually and in the PC analysis. The magnitude of the biases and the details of distribution shapes varied for different VH
germline segments within each group but were consistent across datasets for each germline segment (Supplementary Fig. 3). Germline segments in the same VH subfamily did not always have the
same biases. The range of germline segment prevalence in the various datasets was similar for the different bias groups (Supplementary Fig. 4a). We determined whether the observed
distribution biases were also present in the naive B cell subset. The biases of the Long group were also observed in the naive B cell compartment, without apparent differences relative to
the distributions in the AE compartments (Fig. 2, Supplementary Fig. 2). All the germline segments in the Neutral group showed average CDR H3 length distribution in the naive subset as well.
However, distribution biases of the Short group in the naive compartment were less prominent (Fig. 2 and Supplementary Fig. 2), consistent with the average CDR H3 length analysis (Fig. 1b
and c). Short biases in the naive compartment were mostly limited to the VH3-73 and VH3-15 germline segments in the TX and WA datasets. Despite the differences in overall CDR H3 length
between the TX and WA naive datasets, the biases in the naive compartment had the same trends in both datasets (Fig. 2 and Supplementary Fig. 2). The data analysis was performed with
datasets filtered for sequences likely to belong to the same lineage. To confirm that biases are not due to pockets of clonal expansion, we performed a repertoire similarity index (RSI)
analysis with the CA, TX and MA datasets similar to a recently described method27 (see Methods for more details). Overall, no apparent increase in RSI scores indicating clonal expansion was
associated with regions of positive prevalence biases in different parts of the CDR H3 length spectrum for the different bias groups (Supplementary Fig. 5a), confirming that clonal expansion
does not account for the observed CDR H3 length biases. CDR H3 LENGTH DISTRIBUTION BIASES ARE NOT GENERATED BY RECOMBINATION We next determined whether the biases observed in the naive
compartment are a direct consequence of biases in the VDJ recombination process for each germline segment. For this, we analyzed frameshifted, nonproductive VH sequences that were part of
the naive WA dataset. Nonproductive recombination products are not directly subject to selection and therefore provide information about recombination products prior to any repertoire
selection. As previously reported16, the CDR H3 lengths of nonproductive VH genes are longer than the productively recombined genes in mature B cell subsets (Fig. 2 and Supplementary Fig.
2). However, CDR H3 length for the nonproductive sequences associated with different VH germline segments approximated a Gaussian distribution, with no observable biases associated with
different VH germline segments relative to the overall dataset, except for minor anomalies associated with some germline segments (Fig. 2 and Supplementary Fig. 2). Therefore, heavy chain
recombination mechanisms do not account for the naive repertoire CDR H3 length distribution biases. CDR H3 LENGTH DISTRIBUTION VARIES WITH VL GERMLINE SEGMENT USE We performed a similar
analysis of CDR H3 length distribution as a function of VL germline segment and B cell compartment using PC and visual analysis. Similar to the VH germline segment-associated biases,
VL-associated biases in the AE compartment could be classified into three groups based on the skewness of their distributions, named here “Short” (right-skewed, with high value of PC1),
“Long” (left-skewed, with low value of PC1) and “Neutral” (intermediate values of PC1), present in both the CA and TX datasets, each group including a diverse set of Vκ and Vλ germline
segments (Figs. 3b and 4, top row, and Supplementary Fig. 6). PC1 and PC2 for the light chain were also associated with apparent skewness and kurtosis. The VL Long bias group has Gaussian
CDR H3 length distributions, whereas the VL Short bias group includes distribution shapes with marked deviations from Gaussian, including localized frequency spikes in discrete sections in
the short range. Only Vκ germline segments in the Long group were associated with similar CDR H3 length biases in the TX naive compartment (Fig. 4, Supplementary Fig. 6). The magnitude of
the VL-associated biases varied for different germline segments within each bias group but were consistent between datasets (Supplementary Fig. 7). As above, the RSI analysis results
indicated that clonal expansion does not account for the VL germline segment-associated CDR H3 length biases (Supplementary Fig. 5b). The prevalence of Short group germline segments in the
dataset was lower than for germline segments of the other two groups (Supplementary Fig. 4b). CDR H3 LENGTH IS BIASED AS A FUNCTION OF VH/JH COMBINATION JH germline segments vary in the
number of amino acid residues that can be potentially contributed to CDR H3 from 4 in JH4 to 9 in JH6. We assessed whether differential JH germline segment usage as a function of VH and VL
germline segment use is the basis for V segment-associated CDR H3 length biases. No clear deviations from average JH usage were observed in association with most VH germline segments in the
WA unproductive sequences (Supplementary Fig. 8a and 9, top panel). The observed deviations in JH prevalence do not readily explain CDR H3 distribution biases associated with VH and VL
germline segments (Supplementary Fig. 8b, c and d, and Supplementary Fig. 9, bottom panel) with the exception of VL germline segment Vκ2-28 in the Long CDR H3 bias group, which was
associated with a higher prevalence of the longer JH6 and lower prevalence of the shorter JH4 germline segments in the heavy chain (Supplementary Fig. 8d). We next analyzed CDR H3 length
distributions associated with different VH/JH germline segment combinations, comparing these to the CDR H3 length distribution of all sequences with the corresponding JH germline segment. As
expected, CDR H3 length distributions were generally shifted according to length of the JH segment in the germline regardless of VH germline segment (Fig. 5, Supplementary Fig. 10 and 11).
However, a subset of VH-associated CDR H3 length biases were impacted by JH germline segment in a manner independent of length of the JH segment in the germline, with very similar patterns
in the naive WA and naive SRI subsets (Fig. 5, Supplementary Fig. 10 and 11). These included a short CDR H3 length bias associated with sequences with the VH3-72, VH3-73, and VH3-15 germline
segments combined with the JH5 and/or JH4 germline segments (Fig. 5, Supplementary Fig. 10 and 11). Additional CDR H3 length biases were observed for other VH/JH germline segment
combinations (Supplementary Fig. 10). Our results indicate that CDR H3 length distribution biases are not necessarily uniform for each VH germline segment but may vary in association with JH
germline segment. In addition, the effect of JH on CDR H3 length distribution is not necessarily similar within VH bias groups, indicating some degree of heterogeneity within bias groups.
DIFFERENTIALLY TRIMMED JH SEGMENTS IN THE NAIVE COMPARTMENT The CDR H3 length distribution biases associated with a subset of VH/JH germline segment combinations may be a consequence of
biases in JH trimming as a function of VH germline segment. JH residue occupancy in the last CDR H3 positions of JH4 and JH5 sequences was used to indirectly determine JH trimming. The JH1,
2, 3, and 6 germline segments were not analyzed due to lack of sufficient data or, in the case of JH6, limited CDR H3 length biases associated with it. No apparent biases in JH residue
occupancy relative to the overall dataset was observed for any of the analyzed VH/JH combinations in the nonproductive WA sequences (Supplementary Fig. 12). However, JH residue trimming
biases were observed for different VH/JH combinations in the WA naive compartment (Fig. 6 and Supplementary Fig. 12). General trends in residue occupancy in JH4 were similar in SRI naive
sequences for the VH/JH4 germline segment combinations with sufficient numbers for analysis (Supplementary Fig. 13). Residue-specific trimming biases were mostly coordinated for consecutive
JH residues in each analyzed VH/JH combination, as expected due to the directional nature of trimming. However, closely related VH germline segments can be associated with distinct trimming
biases of different JH4 residues. For instance, VH2-5/JH4 sequences are associated mostly with reduced trimming of IMGT® residue 115 (Phe) whereas for VH2-70/JH4 strongly reduced trimming of
residue 116 (Asp) was also observed (Fig. 5 and Supplementary Fig. 12). The results indicate a complex set of constraints leading to selection of differentially trimmed JH segments in the
context of certain VH and JH germline segments during naive repertoire maturation. DISCUSSION Understanding antibody CDR H3 diversity generation, a process critical for the availability of
immune receptors binding a wide range of antigens, has long been a goal in the immunology and antibody engineering fields. Numerous reports have described overall CDR H3 length and amino
acid composition biases in health and disease and in different B cell developmental stages6,7,8,9,10,11,12,13,14,15,16,17,18,19,20. CDR H3 and junctional segments lengths in the B cell
repertoire have been assumed to be independent of VH and JH germline segments except for their lengths prior to recombination17,19,20. This assumption has been implicitly used, for example,
to generate simulated CDR H3 datasets to estimate the significance of observed clonal convergence in repertoires20. Analysis of the impact of VL on CDR H3 length has been limited to CDR L3
length, with negative results23. Here we describe detailed, high-dimensional analyses of CDR H3 and junctional segment length distributions and show a complex set of biases determined by VH,
VL, and JH germline segment use and B cell maturation state that are not predictable from germline segment lengths and amino acid sequences. Most of the length and junctional biases we
describe are evident in the naive B cell compartment but not in the nonproductive subset, indicating a major role of naive B cell repertoire maturation and, by extension, selection against
self-reactivity or for structural integrity as likely factors in shaping those biases. In addition, only a subset of VH or VL germline segments is associated with biases towards shorter CDR
H3 lengths in the antigen-experienced compartment, indicating general germline segment-specific adaptive immunity selection processes shared among individuals. Similar T cell receptor β
chain CDR3 length distribution biases with different _TRBV_ germline segments in repertoires arise in the process of T cell maturation28, although selective processes may differ between B
and T cell repertoires due to differences in mechanisms of antigen recognition. Our results provide a detailed view into the dynamics of heavy chain junctional biases in antibody repertoires
that complements previously described dynamics of clonal selection and expansion, convergence, sequence diversity and changes in overall CDR H3 properties in B cell
maturation9,10,11,20,22,25,27,29. Special consideration was given to the repeatability and robustness of the findings. The results are based on a total of 12 donors in four datasets and
confirmed by analysis of 8 additional donors from the SRI dataset. These datasets were obtained and parsed with different sequencing methods and bioinformatic pipelines, minimizing the
impact of technical artifacts. Some of the biases, such as those associated with VL, and JH cannot be easily generated by sequencing or parsing artifacts, especially in a systematic fashion
across datasets. The stringency of clonotype clustering criteria had limited impact on results. This is exemplified by the WA and SRI datasets, which yielded CDR H3 length distributions
similar to other datasets (Supplementary Fig. 3) despite having been clustered by clonotype using a higher CDR H3 sequence identity threshold than other datasets (Supplementary Table 1). A
recurring theme in the results presented here was that biases observed at one level (e.g., VH germline segment) were only partly explained by biases at higher-dimensional levels (e.g., VH/JH
combinations), with additional unexpected biases observed in the higher-dimensional levels. It is expected that higher-dimensional analyses including other repertoire descriptors will
uncover additional biases, two examples being DH junctional length biases (Supplementary Fig. 14, 15, and 16 and Supplementary text) and CDR H3 length biases associated with different VH
germline segment allelic variants (Supplementary Fig. 17). Haplotype variations could also potentially affect CDR H3 length distributions in a VH allele-dependent manner or through
differences in DH germline segment composition and differential recombination frequencies of DH or JH germline segments of different lengths in different chromosomes, combined with
differential recombination frequencies of VH alleles30,31,32. However, the observation of essentially the same CDR H3 length distribution biases in several donors from 5 different sources
and junctional segment length biases in 11 donors from 2 of these sources, along with a lack of systematic associations between VH, DH, and JH alleles across donors31,32, indicates that
haplotype variations are unlikely to be a major factor in the CDR H3 and junctional length distribution biases described here. In addition, heavy chain variable region haplotype differences
would not be expected to impact CDR H3 distributions associated with VL germline segments and the AE compartment-specific short CDR H3 length biases. The analyses shown here use germline
segment information as a proxy for undefined sequence features that ultimately determine the observed biases. The selected CDR H3 sequence and structural properties that result in the
observed biases and the germline segment sequence properties that determine those biases remain to be identified. Analysis of VH germline segment residues that can directly encode or bias
the first CDR H3 residues in IMGT® positions 105–107 did not reveal clear correlations between these and most CDR H3 bias groups or junctional segment length biases (Supplementary Fig. 18).
In addition, no obvious correlations between JH trimming biases and variations in VH germline segment residues in positions 40–42 generally contacting the differentially trimmed JH residues
115 and 116 were observed. The differentially trimmed residue 116 is located in a region at the base of CDR H3 that can adopt either a “bulged” or “extended” conformation33,34. The bulged
conformation appears to depend on the Ig domain, encoded mostly by VH germline segments34,35. Whether VH germline segment-dependent JH trimming biases reflect biases in the structure of the
CDR H3 base remains to be determined. One challenge in determining how different germline segment regions determine the observed biases is the relatively limited number of non-redundant
human antibody structures with different VH/JH combinations or VL germline segments with different CDR H3 lengths. The CDR H3 biases described here pose questions about the functional
properties that might shape those biases and the functional consequences of these biases for adaptive immunity. The emergence of some biases in the naive repertoire suggests selection
against self-reactivity, selection for structural integrity, expression or a combination of these factors as possible mechanisms. If related to selection against self-reactivity, the
different biases indicate either that features other than CDR H3 charge and hydrophobicity contribute to self-reactivity or that V germline segments outside CDR H3 modulate the
self-reactivity mediated by these factors. The possible role of selection against self-reactivity may allow leveraging the biases observed in CDR H3 in large natural human antibody
repertoire deep sequencing datasets to understand CDR H3 properties beyond charge that correlate with antibody polyspecificity and self-reactivity, a subject of considerable interest in
therapeutic antibody development and in the understanding of functional consequences of immune disorders with altered CDR H3 sequence profiles11,36,37. The bias towards shorter CDR H3
lengths associated with a subset of VH and VL germline segments in the AE compartment may be attributable to these same mechanisms or to antigen-driven immune selection. The latter would
suggest widespread convergences in human repertoires associated with certain VH and VL germline segments or, possibly, some degree of functional specialization in the germline segment
repertoire linked to short CDR H3 sequences, analogous to the association between CDR H3 length and recognition of different antigen classes38. Our results point to unexpected
cross-constraints between VH, VL, JH, and other junctional elements selected at different stages of B cell development that shape CDR H3 and junctional length distributions in antibody
repertoires. That is, CDR H3 length distribution in the repertoire is not independent of heavy and light chain VH, VL, and VH/JH germline segment usage as implicitly
assumed6,7,8,9,10,11,17,18,19,20. Instead, overall CDR H3 and junctional length distributions in antibody repertoires are aggregates of several sub-repertoires with discrete sets of biases
relative to each other that arise in different stages of B cell maturation as a function of VH, VL, and JH germline segment use, shared by normal donors. Thus, proper description of CDR H3
length biases in disease and immune states11,12,39 requires the context of VH, VL, or VH/JH germline segment usage in which these biases are observed for meaningful interpretation. The
analyses described here provide a high-dimensional CDR H3 analytical framework, in which CDR H3 and junctional length distributions are analyzed in the context of VH, VL, and JH germline
segments and combinations of these, and a baseline of these biases with multiple healthy donors for further studies of B cell repertoire maturation and clonal selection in health and
disease. METHODS DATASETS AND ANALYSIS Sequences were obtained from the original publications14,21,25 except for the MA dataset. The sequences in the MA dataset were obtained from a
re-sequencing by Illumina MiSeq of a set of previously described samples22, deposited in the Sequence Read Archive (SRA) database24. A summary of the samples used here is given in
Supplementary Data 5. Sequencing methods for the MA dataset are described in the experiment design section associated with sample data (see https://www.ncbi.nlm.nih.gov/sra/SRX2251687). The
SRI dataset donors included in the analyses are 316188, 326650, 326737, 326780, 326797, 326907, 327059, and D10320. SRI dataset donors 326713 and 326651 were only used for VH germline
segment allele-specific analyses. Sequences were used as parsed in the original publications except for sequences of the MA dataset obtained from the SRA database24, where the raw sequencing
files were processed and germline segments annotated with a custom pipeline (available from docker hub repository at https://hub.docker.com/r/kamhonhoi/iganalysis). Briefly, paired-end
reads were merged using FLASH40 to reconstruct the full-length variable domain sequences using the following parameters: read length at 300 bps, expected fragment length at 530 bps, standard
deviation at 50 bps. The full-length sequences were subsequently processed to identify the frameworks and CDR regions using position-weighted motifs as previously described41. IgBlast42 was
used to supplement the region parsed data with germline segment annotation for each sequence, including nucleotide somatic mutations. Isotypes of the sequences were determined by finding
the closest matching human CH1 isotypes on the available CH1 sequences. Each sequence was processed and annotated with the frameworks, CDRs, germline segment use and clonotype grouping (see
below). Nonproductive sequences in the WA dataset used for analyses were limited to frameshifted sequences in the naive compartment to minimize the indirect effects of sequencing errors and
clonal expansion. CDR H3 length of nonproductive, frameshifted sequence length in amino acid residues was set as the nearest integer of CDR H3 length in nucleotides divided by 3. For naive
compartment sequences of Donor 1 of the WA dataset only the D1a repeat was used for most analyses25. All CDR H3 length distributions and germline segment prevalence analyses were determined
using custom scripts and Microsoft Excel 2016. The IMGT® CDR definition and numbering system are used throughout26. CLONOTYPE CLUSTERING Clonotypes in the CA and TX datasets were defined as
sequences from the same donor, VH and VL germline segments and CDR H3 length with a nominal 57% or greater CDR H3 amino acid sequence identity, which better approximates an average 60% CDR
H3 sequence identity across the range of CDR H3 lengths. IgG/IgA and IgM sequences were segregated prior to clonotype clustering. For the TX dataset IgG/IgA, CD27pos/IgM and CD27neg
sequences were segregated prior to clonotype clustering. The 11% of sequences without isotype information in the TX AE datasets, which included IgG, IgA, and IgM sequences, were excluded
from the analyses due to the differences between IgG/IgA and IgM overall distributions in the AE compartment (Fig. 1b). Clonotypes in the MA dataset were defined as sequences from the same
donor, VH and JH germline segments and CDR H3 length with a nominal 57% (average 60% identity across CDR H3 lengths) or greater CDR H3 amino acid sequence identity as above. Clonotypes in
the SRI dataset were defined as sequences from the same donor with the same VH and JH germline segment, isotype and same CDR H3 length and sequence. Only sequences labeled as “productive” in
the SRI dataset were analyzed. Only a randomly chosen sequence from each clonotype was retained in the dataset for the TX, CA, WA, MA, and SRI datasets. Clonotypes in the WA dataset were
defined as sequences from the same donor with the same VH and JH germline segment and same CDR H3 length and sequence. If VH germline segment information was not available then VH subfamily
information was used in lieu, retaining as a representative for the clonotype a sequence with VH germline segment information if available. If JH germline segment information was not
available then this parameter was ignored, also retaining otherwise identical sequences with available JH information as representatives for clonotypes, if available. Nonproductive sequences
in the WA dataset were not processed for clonotype clustering. REPERTOIRE SIMILARITY INDEX ANALYSIS RSI was computed in a manner similar to a previously described method27. For a given set
_S_ of CDR H3 sequences, all of the same length _n_, RSI is measured as follows: $$RSI = \left[ {1 - \frac{{{\mathrm{median}}\left\{ {LD\left( {S_i,S_j} \right)_{i \ne j}} \right\}}}{n}}
\right] \times 100\% \,\forall \,1 \le i,j \le |{\boldsymbol{S}}|$$ where _S__j_ and _S__j_ refer to any two sequences in the set of CDR H3 sequences and _LD_(_S__i_,_S__j_) refers to the
Levenshtein distance function, which measures the number of amino acid changes necessary to convert _S__i_ to _S__j_. For a given VH germline segment and CDR H3 length, RSI values were
computed for those sequences that shared the same VL germline segment (for the paired CA and TX datasets) or the same JH germline segment (for the unpaired MA datasets) and the same CDR H3
length. Values were computed separately for each donor in the datasets and averaged for each length. Values shown in graphs in Supplementary Fig. 5 are the averages in each length for
different datasets. All calculations were performed using custom scripts in R. PRINCIPAL COMPONENT ANALYSIS OF CDR H3 LENGTH DISTRIBUTIONS The length distribution of each germline segment
was captured as a vector of length 22 containing the percentage of sequences of length 5–26. For VH, the values for each germline segment were averaged overall the AE datasets except the WA
dataset due to limited germline segment coverage. For VL, the values were averaged over the CA and TX datasets. The distributions of each germline segment were consolidated into a matrix
_X__n_×22 where _n_ is the number of germline segments considered for analysis (_n_ = 39 for VH and _n_ = 35 for VL). The variance covariance matrix _S_22×22 of X was computed with elements
_S__ij_ as $$S_{ij} = < \left( {X - \left\langle {X_i} \right\rangle } \right)\left( {X - \left\langle {X_j} \right\rangle > \forall \,1 \le i,j \le 22} \right.$$ where <> refers
to average across all germline segments. Eigen decomposition of the matrix _S_ results in 22 eigenvectors, each of which capture a trend in the distribution as a function of the CDR H3
lengths and are sorted in decreasing order of the variance they capture. Each germline segment was then projected onto these eigenvectors to obtain the PC scores which enabled visualization
of the different trends and comparisons among the different germline segments. Principal Component Analysis was performed using the ‘princomp’ function and Repertoire Similarity Index,
implemented using the ‘sdists’ function in the package ‘cba’ in R version 3.5.0. STATISTICS AND REPRODUCIBILITY Samples consist of independently sequenced antibody repertoires from 20 donors
from five laboratories under different sequencing conditions, comprising five datasets of with 2–8 donors each with varying number of sequences per donor (see Supplementary Data 1, 2, and 3
for details and sample sizes for each dataset, donor, B cell subset and germline segment). To avoid over-representation of sequences from donors and datasets with higher sequence counts,
calculations were performed per donor and then averaged per dataset or across datasets except for Supplementary Fig. 1, where sequences from donors of each dataset were pooled prior to
calculations. Germline segments within a donor with fewer than 80 counts were excluded from analyses. Reproducibility was assessed by comparing distributions between datasets, with the
exception for nonproductive sequences, which were present only in the WA dataset. Two-tailed paired _t_-tests of CDR H3 length distributions were performed using Microsoft Excel for the Mac
2016. Data from individual donors comparing germline segment-specific to overall repertoire CDR H3 length prevalence from the CA, TX, MA, and WA datasets (_n_ = 11 donors for VH, _n_ = 5
donors for VL) were used for _t-_tests of AE compartment samples over a sliding window of two consecutive CDR H3 lengths to minimize local fluctuations. _P_-value thresholds of 10−4 (VH) and
10−2 (VL) were used to assess significance across the range of CDR H3 lengths. Mann-Whitney tests for distributions in Supplementary Fig. 17 were done using GraphPad Prism version 6.
REPORTING SUMMARY Further information on research design is available in the Nature Research Reporting Summary linked to this article. DATA AVAILABILITY Sequence datasets used in analyses
have been previously described20,21,22,23,24,25. The subset of the datasets with sequences clustered by clonotype used here are available at https://doi.org/10.5061/dryad.cjsxksn2x 43. All
data supporting this study are available within the article and its Supplementary Information Figures and Supplementary Tables or are available from the corresponding author on reasonable
request. CODE AVAILABILITY Raw NGS sequencing data analyzed with a custom analysis pipeline, available from the docker hub repository at hub.docker.com under kamhonhoi/iganalysis. Principal
Component Analysis performed using the ‘princomp’ function and Repertoire Similarity Index, implemented using the ‘sdists’ function in the package ‘cba’ in R version 3.5.0. REFERENCES *
Schroeder, H. W. Jr. & Cavacini, L. Structure and function of immunoglobulins. _J. Allergy Clin. Immunol._ 125, S41–S52 (2010). Article Google Scholar * Cobb, R. M., Oestreich, K. J.,
Osipovich, O. A. & Oltz, E. M. Accessibility control of V(D)J recombination. _Adv. Immunol._ 91, 45–109 (2006). Article CAS Google Scholar * Bassing, C. H., Swat, W. & Alt, F. W.
The mechanism and regulation of chromosomal V(D)J recombination. _Cell_ 109(Suppl), S45–S55 (2002). Article CAS Google Scholar * Rajewsky, K. Clonal selection and learning in the antibody
system. _Nature_ 381, 751–758 (1996). Article CAS Google Scholar * Nemazee, D. Mechanisms of central tolerance for B cells. _Nat. Rev. Immunol._ 17, 281–294 (2017). Article CAS Google
Scholar * Zemlin, M. et al. Expressed murine and human CDR-H3 intervals of equal length exhibit distinct repertoires that differ in their amino acid composition and predicted range of
structures. _J. Mol. Biol._ 334, 733–749 (2003). Article CAS Google Scholar * Schroeder, H. W. Jr. Similarity and divergence in the development and expression of the mouse and human
antibody repertoires. _Dev. Comp. Immunol._ 30, 119–135 (2006). Article CAS Google Scholar * Bagnara, D. et al. A reassessment of IgM memory subsets in humans. _J. Immunol._ 195,
3716–3724 (2015). Article CAS Google Scholar * Wardemann, H. et al. Predominant autoantibody production by early human B cell precursors. _Science_ 301, 1374–1377 (2003). Article CAS
Google Scholar * Wu, Y. C. et al. High-throughput immunoglobulin repertoire analysis distinguishes between human IgM memory and switched memory B-cell populations. _Blood_ 116, 1070–1078
(2010). Article CAS Google Scholar * Roskin, K. M. et al. IgH sequences in common variable immune deficiency reveal altered B cell development and selection. _Sci. Transl. Med._ 7,
302ra135 (2015). Article Google Scholar * Doorenspleet, M. E. et al. Rheumatoid arthritis synovial tissue harbours dominant B-cell and plasma-cell clones associated with autoreactivity.
_Ann. Rheum. Dis._ 73, 756–762 (2014). Article CAS Google Scholar * Bashford-Rogers, R. J. M., Smith, K. G. C. & Thomas, D. C. Antibody repertoire analysis in polygenic autoimmune
diseases. _Immunology_ 155, 3–17 (2018). Article CAS Google Scholar * DeKosky, B. J. et al. Large-scale sequence and structural comparisons of human naive and antigen-experienced antibody
repertoires. _Proc. Natl Acad. Sci. USA_ 113, E2636–E2645 (2016). Article CAS Google Scholar * Ivanov, I. I. et al. Development of the expressed Ig CDR-H3 repertoire is marked by
focusing of constraints in length, amino acid use, and charge that are first established in early B cell progenitors. _J. Immunol._ 174, 7773–7780 (2005). Article CAS Google Scholar *
Larimore, K., McCormick, M. W., Robins, H. S. & Greenberg, P. D. Shaping of human germline IgH repertoires revealed by deep sequencing. _J. Immunol._ 189, 3221–3230 (2012). Article CAS
Google Scholar * Elhanati, Y. et al. Inferring processes underlying B-cell repertoire diversity. _Philos. Trans. R. Soc. Lond. B Biol. Sci._ 370, 20140243,
https://doi.org/10.1098/rstb.2014.0243 (2015). Article CAS PubMed PubMed Central Google Scholar * Miqueu, P. et al. Statistical analysis of CDR3 length distributions for the assessment
of T and B cell repertoire biases. _Mol. Immunol._ 44, 1057–1064 (2007). Article CAS Google Scholar * Marcou, Q., Mora, T. & Walczak, A. M. High-throughput immune repertoire analysis
with IGoR. _Nat. Commun._ 9, 561 (2018). Article Google Scholar * Briney, B., Inderbitzin, A., Joyce, C. & Burton, D. R. Commonality despite exceptional diversity in the baseline human
antibody repertoire. _Nature_ 566, 393–397 (2019). Article Google Scholar * Goldstein, L. D. et al. Massively parallel single-cell B-cell receptor sequencing enables rapid discovery of
diverse antigen-reactive antibodies. _Commun. Biol._ 2, 304 (2019). Article Google Scholar * Laserson, U. et al. High-resolution antibody dynamics of vaccine-induced immune responses.
_Proc. Natl Acad. Sci. USA_ 111, 4928–4933 (2014). Article CAS Google Scholar * DeKosky, B. J. et al. In-depth determination and analysis of the human paired heavy- and light-chain
antibody repertoire. _Nat. Med._ 21, 86–91 (2015). Article CAS Google Scholar * Busse, C. Dynamics of the human antibody repertoire after influenza vaccination. _NCBI BioProject
Database_, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA349143 (2016). * DeWitt, W. S. et al. A public database of memory and naive B-cell receptor sequences. _PLoS ONE_ 11, e0160853 (2016).
Article Google Scholar * Lefranc, M. P. IMGT, the international ImMunoGeneTics database: a high-quality information system for comparative immunogenetics and immunology. _Dev. Comp.
Immunol._ 26, 697–705 (2002). Article CAS Google Scholar * Greiff, V. et al. Systems analysis reveals high genetic and antigen-driven predetermination of antibody repertoires throughout B
cell development. _Cell Rep._ 19, 1467–1478 (2017). Article CAS Google Scholar * Matsutani, T. et al. Comparison of CDR3 length among thymocyte subpopulations: impacts of MHC and BV
segment on the CDR3 shortening. _Mol. Immunol._ 44, 2378–2387 (2007). Article CAS Google Scholar * Miho, E., Roskar, R., Greiff, V. & Reddy, S. T. Large-scale network analysis reveals
the sequence space architecture of antibody repertoires. _Nat. Commun._ 10, 1321 (2019). Article Google Scholar * Boyd, S. D. et al. Individual variation in the germline Ig gene
repertoire inferred from variable region gene rearrangements. _J. Immunol._ 184, 6986–6992 (2010). Article CAS Google Scholar * Kidd, M. J. et al. The inference of phased haplotypes for
the immunoglobulin H chain V region gene loci by analysis of VDJ gene rearrangements. _J. Immunol._ 188, 1333–1340 (2012). Article CAS Google Scholar * Gidoni, M. et al. Mosaic deletion
patterns of the human antibody heavy chain gene locus shown by Bayesian haplotyping. _Nat. Commun._ 10, 628 (2019). Article CAS Google Scholar * Shirai, H., Kidera, A. & Nakamura, H.
H3-rules: identification of CDR-H3 structures in antibodies. _FEBS Lett._ 455, 188–197 (1999). Article CAS Google Scholar * North, B., Lehmann, A. & Dunbrack, R. L. Jr. A new
clustering of antibody CDR loop conformations. _J. Mol. Biol._ 406, 228–256 (2011). Article CAS Google Scholar * Weitzner, B. D., Dunbrack, R. L. Jr. & Gray, J. J. The origin of CDR
H3 structural diversity. _Structure_ 23, 302–311 (2015). Article CAS Google Scholar * Vander Heiden, J. A. et al. Dysregulation of B cell repertoire formation in Myasthenia Gravis
patients revealed through deep sequencing. _J. Immunol._ 198, 1460–1473 (2017). Article Google Scholar * Jain, T. et al. Biophysical properties of the clinical-stage antibody landscape.
_Proc. Natl Acad. Sci. USA_ 114, 944–949 (2017). Article CAS Google Scholar * Collis, A. V., Brouwer, A. P. & Martin, A. C. Analysis of the antigen combining site: correlations
between length and sequence composition of the hypervariable loops and the nature of the antigen. _J. Mol. Biol._ 325, 337–354 (2003). Article CAS Google Scholar * Breden, F. et al.
Comparison of antibody repertoires produced by HIV-1 infection, other chronic and acute infections, and systemic autoimmune disease. _PLoS ONE_ 6, e16857 (2011). Article CAS Google Scholar
* Magoc, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. _Bioinformatics_ 27, 2957–2963 (2011). Article CAS Google Scholar * Chen, Y.
et al. Barcoded sequencing workflow for high throughput digitization of hybridoma antibody variable domain sequences. _J. Immunol. Methods_ 455, 88–94 (2018). Article CAS Google Scholar
* Ye, J., Ma, N., Madden, T. L. & Ostell, J. M. IgBLAST: an immunoglobulin variable domain sequence analysis tool. _Nucleic Acids Res._ 41, W34–W40 (2013). Article Google Scholar *
Sankar, K., Hoi, K. H., Jr. & Hotzel, I. Dynamics of heavy chain junctional length biases in antibody repertoires. _Dryad Database_, https://doi.org/10.5061/dryad.cjsxksn5062x (2020).
Download references ACKNOWLEDGEMENTS We thank Steve Guerrero and Qing Zhang for helpful discussions. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Antibody Engineering, South
San Francisco, CA, 94080, USA Kannan Sankar, Kam Hon Hoi & Isidro Hötzel * Department of Bioinformatics and Computational Biology, Genentech, South San Francisco, CA, 94080, USA Kam Hon
Hoi Authors * Kannan Sankar View author publications You can also search for this author inPubMed Google Scholar * Kam Hon Hoi View author publications You can also search for this author
inPubMed Google Scholar * Isidro Hötzel View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Conceptualization: I.H.; Formal analysis: I.H.,
K.S.; Investigation: I.H., K.S.; Methodology: I.H., K.S., K.H.H.; Supervision: .I.H.; Writing—original draft: I.H., K.S.; Writing—review and editing: I.H., K.S., K.H.H. Based on CRediT
contributor taxonomy. CORRESPONDING AUTHOR Correspondence to Isidro Hötzel. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing non-financial interests but the following
competing financial interests: the authors are current or past Genentech employees and may hold shares in Roche. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with
regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION SUPPLEMENTARY DATA 1-5 SUPPLEMENTARY DATA 6 REPORTING
SUMMARY DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which
permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to
the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless
indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or
exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints
and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Sankar, K., Hoi, K.H. & Hötzel, I. Dynamics of heavy chain junctional length biases in antibody repertoires. _Commun Biol_ 3, 207
(2020). https://doi.org/10.1038/s42003-020-0931-3 Download citation * Received: 05 August 2019 * Accepted: 01 April 2020 * Published: 01 May 2020 * DOI:
https://doi.org/10.1038/s42003-020-0931-3 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative