Loss of soluble guanylyl cyclase in platelets contributes to atherosclerotic plaque formation and vascular inflammation
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Variants in genes encoding the soluble guanylyl cyclase (sGC) in platelets are associated with coronary artery disease (CAD) risk. Here, by using histology, flow cytometry and
intravital microscopy, we show that functional loss of sGC in platelets of atherosclerosis-prone _Ldlr_−/− mice contributes to atherosclerotic plaque formation, particularly via increasing
in vivo leukocyte adhesion to atherosclerotic lesions. In vitro experiments revealed that supernatant from activated platelets lacking sGC promotes leukocyte adhesion to endothelial cells
(ECs) by activating ECs. Profiling of platelet-released cytokines indicated that reduced platelet angiopoietin-1 release by sGC-depleted platelets, which was validated in isolated human
platelets from carriers of _GUCY1A1_ risk alleles, enhances leukocyte adhesion to ECs. Importantly, pharmacological sGC stimulation increased platelet angiopoietin-1 release in vitro and
reduced leukocyte recruitment and atherosclerotic plaque formation in atherosclerosis-prone _Ldlr_−/− mice. Therefore, pharmacological sGC stimulation might represent a potential therapeutic
strategy to prevent and treat CAD. SIMILAR CONTENT BEING VIEWED BY OTHERS SVEP1 IS AN ENDOGENOUS LIGAND FOR THE ORPHAN RECEPTOR PEAR1 Article Open access 15 February 2023 MILD
HYPERLIPIDEMIA IN MICE AGGRAVATES PLATELET RESPONSIVENESS IN THROMBUS FORMATION AND EXPLORATION OF PLATELET PROTEOME AND LIPIDOME Article Open access 08 December 2020 ACKR3 REGULATES
PLATELET ACTIVATION AND ISCHEMIA-REPERFUSION TISSUE INJURY Article Open access 05 April 2022 MAIN CAD is a leading cause of morbidity and mortality in industrialized nations1. In the past 15
years, large-scale genetic studies led to the identification of more than 300 genomic variants that are associated with CAD and myocardial infarction (MI) risk. Interestingly, most of the
affected genes do not influence traditional risk factors2 but rather mechanisms not previously implicated in the pathophysiology of atherosclerosis. One prominent example is the nitric oxide
(NO)–cyclic guanosine monophosphate (cGMP) signaling pathway, which contains several genome-wide significantly associated variants for CAD3. In the center of this pathway sits sGC, which is
activated by NO and produces the second messenger cGMP. From a genetic perspective, private mutations in the genes encoding for sGC and rare coding and common noncoding variants in the
_GUCY1A1_ gene, which encodes the α1-subunit of the sGC, were all associated with CAD or premature MI by exome sequencing and genome-wide association studies (GWAS), respectively4,5. These
mutations and variants reduce sGC expression or activity5,6,7; in line with this, enhanced NO–cGMP signaling has been associated with reduced risk of several cardiometabolic phenotypes
including CAD and peripheral artery disease8. _GUCY1A1_ and _GUCY1B1_, genes encoding sGC, are expressed at high levels in platelets and vascular smooth muscle cells. In humans, carriers of
the CAD risk variant rs7692387 have reduced sGC α1 protein levels, which might impair the effects of the natural platelet inhibitor NO7. Indeed, a retrospective analysis of two randomized
trials revealed that inhibition of platelet activity by aspirin successfully reduced cardiovascular events in primary prevention only in homozygous carriers of the _GUCY1A1_ risk allele9.
Since the overall role of platelets in atherosclerosis is controversial10, we decided to delete sGC in mice platelets specifically by knockout of its β1-subunit to investigate the
contribution of platelet sGC on atherosclerosis and vascular inflammation and further evaluate the potential of sGC stimulation as a therapeutic strategy. RESULTS ATHEROSCLEROTIC PLAQUE
FORMATION IN MICE LACKING PLATELET SGC We created mice with a platelet-predominant sGC knockout (Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP_) which displayed reduced sGC β1 protein levels compared to
wild-type (WT) platelet sGC mice (Extended Data Fig. 1a); expectedly, this reduced inhibition of agonist-induced platelet aggregation by the NO donor sodium nitroprusside (Extended Data Fig.
2). Importantly, the expression of other sGC subunits was not altered (Extended Data Fig. 1b,c). To examine the effect of platelet sGC on atherosclerosis, we crossbred these animals with
atherosclerosis-prone mice (_Ldlr_−/− mice) which were then fed a Western diet for ten weeks. Body weight, serum cholesterol, and hematological parameters were comparable between the
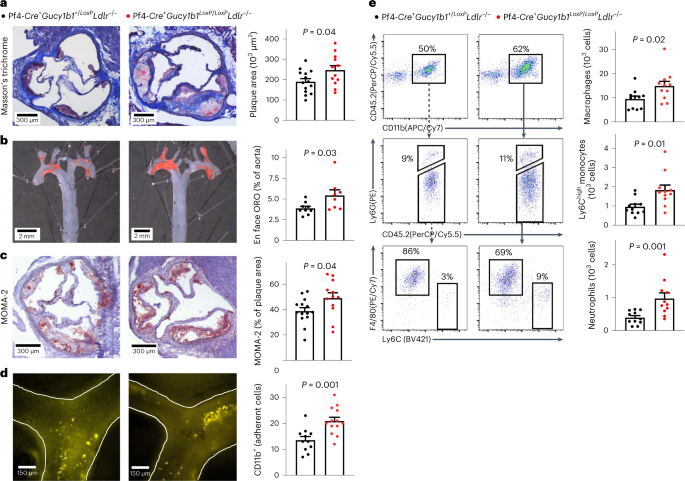
genotypes (Extended Data Fig. 3). Pf4-_Cre_+_Gucy1b1__L__oxP_/_LoxP__Ldlr_−/− mice displayed larger plaques in the aortic root (246,998 ± 22,162 µm2 (_n_ = 12) versus 189,843 ± 15,156 µm2
(_n_ = 14), _P_ = 0.04; Fig. 1a) and aorta en face analysis (5.4 ± 0.7% (_n_ = 8) versus 3.9 ± 0.2% (_n_ = 9), _P_ = 0.03; Fig. 1b). Immunohistochemical staining of aortic root sections
revealed increased plaque monocyte and macrophage content in Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP__Ldlr_−/− mice compared to Pf4-_Cre_+_Gucy1b1_+/_LoxP__Ldlr_−/− mice (49.1 ± 4.2% (_n_ = 12)
versus 38.8 ± 2.8% (_n_ = 14) of plaque area, _P_ = 0.04; Fig. 1c). To explore why monocyte/macrophages accumulated more in mice with platelet-predominant sGC depletion, we performed
intravital fluorescence microscopy of carotid artery plaques in Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP__Ldlr_−/− and Pf4-Cre+_Gucy1b1_+/_LoxP__Ldlr_−/− mice which were fed a Western diet for six
weeks to initiate plaque formation. We found enhanced adhesion of leukocytes in Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP__Ldlr_−/− compared to Pf4-_Cre_+_Gucy1b1_+/_LoxP__Ldlr_−/− mice (20.9 ± 1.5
(_n_ = 13) versus 13.6 ± 1.4 (_n_ = 11) Cd11b+ cells, _P_ = 0.001; Fig. 1d and Extended Data Fig. 4). Flow cytometry of aortic cell suspensions revealed more numerous Ly6Chigh monocytes
(1,829 ± 262 versus 964 ± 129 cells per aorta, _n_ = 11 each, _P_ = 0.01), neutrophils (974 ± 170 versus 392 ± 60 cells per aorta, _n_ = 11 each, _P_ = 0.001) and macrophages (14,895 ± 1,912
versus 9,463 ± 1,211 cells per aorta, _n_ = 11 each, _P_ = 0.02; Fig. 1e) in Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP__Ldlr_−/− compared to Pf4-_Cre_+_Gucy1b1_+/_LoxP__Ldlr_−/− mice while blood
leukocyte numbers were unchanged (Extended Data Fig. 3d). Taken together, these data indicate that the lack of sGC in platelets contributes to vascular inflammation and hence atherosclerotic
plaque progression. PLATELET SGC AND LEUKOCYTE ADHESION IN VITRO To follow up on enhanced adhesion of leukocytes to atherosclerotic plaques in mice lacking platelet sGC under proatherogenic
conditions, we tested whether this phenotype can be resembled in vitro. Therefore, we isolated blood monocytes and neutrophils from WT mice and incubated these with WT aortic ECs in the
presence of activated platelet releasate from Pf4-_Cre_+_Gucy1b1_+/_LoxP_ or Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP_ mice. We found that incubation with the supernatant of activated platelets
lacking sGC enhanced leukocyte, particularly monocyte (33,125 ± 1,313 versus 27,039 ± 555 relative fluorescence units (RFU), _P_ = 0.006, _n_ = 8; Fig. 2a) and neutrophil (33,810 ± 1,139
versus 27,824 ± 758 RFU, _P_ < 0.001, _n_ = 8; Fig. 2b) adhesion. To delineate whether leukocytes or ECs are activated by the sGC knockout platelet releasate, we preincubated neutrophils
or monocytes and ECs with activated platelet supernatant from Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP_ mice before performing the adhesion assay. We found that preincubation of ECs with supernatant
from activated sGC knockout platelets increased adhesion compared to preincubation of neutrophils (31,383 ± 1,731 versus 22,254 ± 1,662 RFU, _P_adj < 0.001, _n_ = 12 experiments; Fig. 2c)
or monocytes (Extended Data Fig. 5). In line with this, already at this very early time point, we found enhanced expression of the adhesion molecule _Vcam1_ in ECs that were incubated with
supernatant of activated Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP_ platelets (Extended Data Fig. 6). These data indicate (1) that platelets from Pf4-_Cre_+_Gucy1b1_+/_LoxP_ and
Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP_ mice differentially release a soluble factor and (2) that this preferentially leads to activation of ECs. REDUCED RELEASE OF ANGIOPOIETIN-1 BY SGC-DEFICIENT
PLATELETS We observed the influence of a soluble factor released by platelets on leukocyte adhesion to ECs. To identify such factors, we next performed cytokine profiling with supernatant of
activated Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP_ and Pf4-_Cre_+_Gucy1b1_+/_LoxP_ platelets. Signal intensity analysis revealed lower angiopoietin-1 (ANGPT1) levels in the supernatant from
Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP_ platelets (3.2 ± 0.4 (_n_ = 7) versus 6.7 ± 0.8 (_n_ = 8) arbitrary units (a.u.), _P_ = 0.002; Fig. 3a and Extended Data Fig. 7). We next aimed at
replicating this finding in an independent cohort of mice using an enzyme-linked immunosorbent assay (ELISA). Importantly, ANGPT1 levels were comparable in quiescent platelets (0.21 ± 0.01
versus 0.23 ± 0.01 pg 10−3 platelets, _n_ = 6 each, _P_ = 0.33; Fig. 3b) as well as in platelet-poor plasma (PPP) from Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP_ and Pf4-_Cre_+_Gucy1b1_+/_LoxP_ mice
(0.6 ± 0.2 ng ml−1 (_n_ = 6) versus 0.4 ± 0.1 ml−1 (_n_ = 5), _P_ = 0.46; Fig. 3c). In line with the explorative analysis displayed in Fig. 3a, we found Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP_
platelets to release reduced amounts of ANGPT1 on activation (30.4 ± 6.4 versus 60.3 ± 4.7 ng ml−1, _n_ = 6 each, _P_ = 0.004; Fig. 3d). ANGPT1 decreases particularly vascular endothelial
growth factor (VEGF)-mediated adhesion of leukocytes to ECs11 and binds to the Tie2 receptor on ECs12. We used the Tie2 inhibitor BAY-826 to investigate whether Tie2 inhibition influences
leukocyte adhesion and found a 17% (±1.2%, _n_ = 11 experiments, _P_ = 0.04) increase in leukocyte adhesion secondary to the inhibition of the ANGPT1 receptor (Fig. 3e). These data
demonstrate that ANGPT1 represents a candidate for mediating the effects of platelet sGC on leukocyte recruitment. Of note, inositol 1,4,5-trisphosphate receptor-associated cGMP-kinase
substrate (IRAG), which represents a downstream effector of cGMP specifically in modulating platelet activity, is encoded by the _IRAG1_ (ref. 13) (previously _IRAG_ or as human homolog
_MRVI1_) gene and has also been associated with CAD by GWAS14. To investigate whether differential ANGPT1 release is mediated via IRAG, we generated Pf4-_Cre_+_Irag1__LoxP_/_LoxP_ mice and
investigated platelet ANGPT1 release compared to the respective controls. In contrast to Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP_ platelets, we did not detect a difference between the genotypes in
this experiment indicating that the influence of sGC on platelet ANGPT1 release is independent of IRAG (Extended Data Fig. 8). It was previously shown that the genotype of the rs7692387 CAD
risk allele at the _GUCY1A1_ locus4 influences _GUCY1A1_ expression in different tissues8 and sGC α1 protein levels in platelets in particular7. Therefore, we tested whether the rs7692387
genotype is associated with ANGPT1 release from platelets in healthy human individuals (_n_ = 5 each; for characteristics see Supplementary Table 3) and found that homozygous carriers of the
CAD risk allele G display lower ANGPT1 release compared to heterozygous or homozygous carriers of the non-risk allele (4.5 ± 0.7 versus 8.3 ± 1.4 ng ml−1, _P_ = 0.04; Fig. 3f). To explore
the role of ANGPT1 in relation to the genes encoding sGC in humans, we queried the STARNET database which contains bulk RNA sequencing (RNA-seq) data from seven cardiometabolic tissues from
patients undergoing coronary artery bypass graft surgery15,16. _ANGPT1_ was detected per tissue in seven distinct coexpression modules (Fig. 3g). Of note, _ANGPT1_ was coexpressed with both
_GUCY1A1_ and _GUCY1B1_ (encoding sGC α1 and sGC β1) in all tissues except mammary artery represented by coexpression module 63 (Fig. 3g). These data suggest ubiquitous presence and clinical
variation in the amounts of platelets. To investigate circulatory rather than tissue-resident platelets, we further analyzed coexpression module 11 from whole-blood samples; this module
consisted of 1,016 genes and was estimated to account for 4.9% of CAD heritability by considering expression quantitative trait locus (eQTL) genes in a meta-analysis of nine GWAS using the
restricted maximum likelihood method17. Coexpression analysis revealed positive correlation between _GUCY1A1_/_GUCY1B1_ and _ANGPT1_ expression and enrichment for genes involved in the Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway platelet activation (Fig. 3h). Altogether this suggests that a substantial proportion of CAD heritability could be mediated by platelets.
Furthermore, Gene Ontology (GO) enrichment analysis of this module (Supplementary Table 4) revealed, among others, ‘response to wounding’ (_P_ = 4.71 × 10−63), ‘wound healing’ (_P_ = 1.84 ×
10−60), blood coagulation (_P_ = 1.99 × 10−48) and ‘regulation of locomotion’ (_P_ = 6.64 × 10−44). These findings support the role of platelets and the interaction of ANGPT1 with sGC in
human CAD. MODULATION OF ANGPT1 RELEASE BY SGC STIMULATION The sGC represents a druggable target and sGC stimulators are used in different clinical scenarios, for example, riociguat in
pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension18 or vericiguat in chronic heart failure19. We next aimed at investigating whether modulation of sGC using a
vericiguat-like sGC stimulator (BAY-747) can influence the release of ANGPT1 and vascular inflammation. To this end, we first incubated platelets from WT mice with BAY-747 or vehicle and
analyzed ANGPT1 release and leukocyte adhesion to ECs. Of note, the platelets of Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP_ mice are not responsive to BAY-747 regarding, for example, platelet
aggregation (Extended Data Fig. 9). Stimulation of sGC with BAY-747 in WT platelets doubled ANGPT1 release (133.8 ± 6.5 ng ml−1 versus 64.0 ± 4.3 ng ml−1, _n_ = 4 each, _P_ = 0.002; Fig. 4a)
and reduced neutrophil adhesion to ECs by 15.6% (± 6.5%, _n_ = 7 each, _P_ = 0.02; Fig. 4b). To determine which downstream cGMP pathways instead of IRAG could influence ANGPT1 release on
platelet activation by shaking, we performed serine/threonine kinase profiling in WT platelets that were preincubated with BAY-747 and then activated by shaking. As displayed in
Supplementary Table 5, sGC stimulation led to a marked increase in the activation of kinases from the AGC group, which include protein kinase C (PKC) and protein kinase G (PKG). To determine
whether the PKC pathway is involved in ANGPT1 release and whether canonical cGMP signaling is involved in its regulation, we next inhibited the inositol 1,4,5-trisphosphate receptor
(IP3-R), PKC and PKG directly and measured ANGPT1 release secondary to activation by shaking (Fig. 4c). Compared to vehicle, inhibition of IP3-R (−23.8 ± 2.8 ng ml−1, _n_ = 5, _P_adj =
0.01), PKC (−23.9 ± 1.4 ng ml−1, _n_ = 6, _P_adj < 0.001) and PKG (−21.3 ± 3.5 ng ml−1, _n_ = 6, _P_adj = 0.004) reduced ANGPT1 release. In contrast, inhibition of further downstream
pathways did not result in altered ANGPT1 release (Fig. 4d). In an analysis of phosphorylated peptides, main targets of NO–cGMP signaling were detected, including endothelial NO synthase and
vasodilator-stimulated phosphoprotein being among the top ten phosphorylated targets (Supplementary Table 6). These data indicate that ANGPT1 release by platelets induced by shaking is
mediated via the PKC pathway and modulated by cGMP via canonical cGMP signaling. THERAPEUTIC POTENTIAL OF STIMULATING SGC IN ATHEROSCLEROSIS To investigate whether sGC stimulation in vivo
can reduce recruitment from blood to the vascular wall, we fed _Ldlr_−/− mice a Western diet containing 0 or 150 parts per million (ppm) BAY-747 and adoptively transferred green fluorescent
protein (GFP)+ myeloid cells after 6 weeks. We retrieved GFP+ monocytes admixed with neutrophils from naïve transgenic Ubc-GFP mice (all leukocytes express GFP+) and injected the cells
intravenously into _Ldlr__−/−_ mice (all cells are GFP−). After a further 24 h, mice were killed and the numbers of GFP+ cells were determined using flow cytometry of aortic cell
suspensions. Mice that received the Western diet containing the sGC stimulator displayed reduced numbers of aortic GFP+ cells (21.4 ± 3.3 versus 42.8 ± 6.9 cells, _n_ = 8 each, _P_ = 0.01;
Fig. 5a). These data and the data displayed in Fig. 4 indicate that pharmacological sGC stimulation can modulate platelet ANGPT1 release, in vitro leukocyte adhesion, and in vivo leukocyte
recruitment. To determine whether such treatment can reduce atherosclerotic plaque formation, we again fed atherosclerosis-prone _Ldlr_−/− mice a Western diet for ten weeks. In one group,
the diet contained 150 ppm BAY-747 (treatment group) while the other group received a Western diet without (0 ppm) BAY-747 (control group). Both groups had elevated serum cholesterol levels
without significant difference between the two groups; similarly, platelet count, blood leukocytes and body weight after the diet were comparable (Extended Data Fig. 10a–d). Under
steady-state conditions, BAY-747 plasma levels in male and female mice receiving BAY-747 were 61.7 ± 1.4 µg l−1 and 40.3 ± 1.7 µg l−1 (_n_ = 6, each), respectively; in the control group,
BAY-747 was not detectable in plasma as expected (Extended Data Fig. 10e). In the aortic root, we found significantly reduced atherosclerotic plaque formation in mice of the treatment group
(62.5 ± 16.1 µm2 (_n_ = 7) versus 123.1 ± 18.6 µm2 (_n_ = 9), _P_ = 0.03; Fig. 5b). Furthermore, we detected fewer leukocytes in the aortic roots of those animals (30.3 ± 5.8% (_n_ = 8)
versus 50.5 ± 6.3% (_n_ = 11) of plaque area, _P_ = 0.04; Fig. 5c). In cell suspensions of the whole aorta, we found fewer numerous macrophages (13,915 ± 1,550 versus 22,156 ± 2,737, _n_ =
12 each, _P_ = 0.02; Fig. 5d), Ly6Chigh monocytes (1,485 ± 348 versus 2,688 ± 531, _n_ = 12 each, _P_ = 0.07; Fig. 5d) and neutrophils (1,201 ± 192 (_n_ = 11) versus 1,885 ± 232 (_n_ = 12),
_P_ = 0.04; Fig. 5d) per aorta in mice treated with the sGC stimulator compared to mice receiving the control diet. Blood leukocyte numbers as determined by flow cytometry and leukocyte
subsets were comparable between both groups (Extended Data Fig. 10f). Taken together, these data indicate that pharmacological sGC stimulation reduces atherosclerotic plaque formation and
vascular inflammation via dampened recruitment of inflammatory leukocytes from blood to plaque. DISCUSSION NO–sGC–cGMP signaling has important functions in several cell types. For instance,
increasing intracellular cGMP levels inhibits migration of vascular smooth muscle cells and aggregation of platelets, respectively20,21. The observation that genes encoding key proteins in
this pathway were associated with CAD and premature MI by GWAS4,14,22,23 and exome sequencing studies5,8,24 renders an important role in the pathophysiology of coronary atherosclerosis
likely. In this study, we sought to specifically investigate the role of platelet sGC in atherosclerosis because we found that carriers of the common, noncoding risk variant rs7692387
identified by GWAS4 displayed reduced expression of sGC in platelets and, as a consequence, reduced inhibition of platelet aggregation on NO stimulation7. In addition, we observed that in
contrast to the general population, individuals who are homozygous for this variant had a benefit from aspirin treatment in primary prevention of cardiovascular events9. In a series of in
vivo and in vitro experiments, we observed larger atherosclerotic plaques in the aortic roots of mice lacking platelet sGC compared to sGC WT mice. Platelet sGC has just recently been
described to act as an endogenous brake on platelet aggregation25. Indeed, it has been hypothesized that platelets adhering to endothelial or plaque erosions may be activated more or less
depending of the availability of sGC. This may stimulate inflammation locally and, subsequently, facilitate atherosclerotic plaque formation. While this is a hypothesis, we report evidence
that platelet sGC influences the release of the soluble factor ANGPT1 with reduced amounts released by platelets lacking sGC. The role of ANGPT1 in atherosclerosis is controversial since
there are reports describing a protective11,12,26 and a deleterious role27. However, given the cellular findings describing the inhibitory effect of ANGPT1 on leukocyte adhesion and vascular
atherosclerosis reported in the literature11, which were confirmed in this study, and the beneficial role of ANGPT1–Tie2 signaling in inflammatory diseases in general28, it represents an
interesting candidate mediating the downstream effects of platelet sGC signaling. In addition, its effects might be depending on coreleased factors. Importantly, the role of sGC in platelets
exceeds the role as a brake on aggregation: while sGC passivates platelet aggregation, it enhances the release of ANGPT1 when platelets are modestly activated. The notion that this effect
is independent of aggregation is further supported by the findings that (1) IRAG, the mediator of cGMP-dependent inhibition of platelet aggregation29 was not significantly involved in ANGPT1
release and (2) that ANGPT1 release was induced by shaking but not, for example, ADP or arachidonic acid. ANGPT1 release rather seems to be mediated by PKC activity and modulated via
canonical cGMP signaling. It is important to acknowledge that the content of ANGPT1 in WT and knockout platelets was similar further indicating a direct link between sGC availability and
ANGPT1 release. This is further emphasized by the finding that a genetically determined reduction, but not lack of, sGC in the platelets of carriers of the CAD-associated risk variant
rs7692387 was associated with a reduction in ANGPT1 release. This, on the one hand, supports the translational relevance of this by nature artificial in vitro and animal studies; on the
other hand, it raises the question whether modulators of sGC function could be used to prevent and treat coronary atherosclerosis. Indeed, stimulators of sGC are emerging as therapeutic
compounds for different cardiovascular diseases (for an overview see Sandner et al.30). Recently, the sGC stimulator vericiguat was found to lower the risk of death from cardiovascular
causes or hospitalization for heart failure in patients with heart failure with reduced ejection fraction19. However, data on atherosclerotic plaque formation and ischemic cardiovascular
events are lacking thus far. To this end, we investigated whether sGC stimulation using a vericiguat-like stimulator, BAY-747, can modulate the reported cellular and phenotypic effects.
First, we found that sGC stimulation can increase ANGPT1 release and subsequently reduce leukocyte adhesion to ECs in vitro. Since we postulate that sGC contributes to reducing vascular
inflammation, we next studied whether sGC stimulation can alter the recruitment of inflammatory leukocytes from blood to plaque. In an adoptive transfer experiment, we observed a reduction
in leukocyte recruitment that is furthermore in line with previous reports showing anti-inflammatory effects of cGMP-increasing pharmacological compounds31,32. The finding of multiple
genome-wide significant hits in genes that encode proteins tightly involved in the formation (_NOS3_ (ref. 22), _GUCY1A1_ (ref. 4)), fate (_PDE5A_23,24), or mediating the downstream effects
of cGMP (_IRAG1_ (ref. 14), _PDE3A_33) and the observation that sGC levels are reduced in atherosclerotic tissues34 increase the likelihood that targeting sGC might be beneficial in CAD. In
atherosclerosis-prone mice on a Western diet, stimulating sGC with BAY-747 led to a reduction in atherosclerotic plaque formation and vascular inflammation warranting further investigation
into this promising pharmacological treatment strategy. Further evidence might be taken from a recent study that compared two pharmacological strategies which are used to treat erectile
dysfunction: compared to alprostadil, that is, prostaglandin E1, treatment with the inhibitor of phosphodiesterase 5A, sildenafil, was associated with a reduced risk of all-cause mortality
and MI in men suffering from CAD35. However, to definitively prove a beneficial role in CAD, prospective clinical trial data are needed. Taken together, we provide further evidence for a
crucial role of platelets in atherosclerosis in general, and of platelet sGC in particular. As shown by our in vitro studies using human and murine biospecimen and in vivo studies, we
postulate an endogenous inhibitory role of platelet sGC on EC-mediated leukocyte recruitment (Fig. 6). We are aware that platelets are not the only cell type in which sGC activity and
genetic variants modulating its availability influence CAD risk. Modulating sGC activity, especially using stimulators, might nevertheless be a promising therapeutic strategy exceeding the
effects on platelet sGC. Our study has several limitations. First, this is an in vitro and mouse in vivo study that cannot resemble the human physiology and pathophysiology. However, the
finding that lack of platelet sGC in mice and genetically determined reduced platelet sGC α1 in humans both reduce release of platelet ANGPT1 supports a possible translation. Furthermore,
similar to humans, a genetically determined reduction in _Gucy1a1_ messenger RNA was associated with increased atherosclerotic plaque formation in the hybrid mouse diversity panel7,36.
Second, although we have shown that reduced ANGPT1 release and enhanced leukocyte adhesion to ECs is linked to reduced or lacking platelet sGC availability and that stimulation of sGC can
modulate these downstream effects, the exact molecular mechanism linking sGC and ANGPT1 release is to be explored. While it is well known that platelets contain at least three types of
secretory granules, with α-granules that also harbor ANGPT1 (ref. 37) being the most abundant type, there is also evidence for the existence of functionally distinct subpopulations within
α-granules38, which may allow selective release of their contents by different stimuli39,40. A similar observation has been reported in neutrophils regarding storage of ANGPT1 and VEGF41. We
further provide evidence that it is independent of IRAG but mediated via canonical cGMP signaling. Third, we know that ANGPT1 is likely not the only mediator of sGC effects on
atherosclerosis. Rather, ANGPT1 might modulate the effect of other cytokines and mediators that are released by platelets on modest activation. Fourth, although we show that platelet sGC
activity influences leukocyte recruitment and atherosclerotic plaque formation, the benefit of systemic sGC stimulators might be influenced by the effects on sGC in other cell types, for
example, vascular smooth muscle cells. Lastly, we cannot generalize a benefit of sGC stimulation to human platelets. Yet our study can, in addition to its biological implications, be
regarded as hypothesis-generating for future clinical trials investigating whether modulating cGMP formation is useful in addition to reduce, for example, low-density lipoprotein cholesterol
levels and residual inflammatory risk. METHODS MOUSE STUDIES Animal experiments were conducted in accordance with the German legislation on the protection of animals and approved by the
local animal care committee (District Government of Upper Bavaria, GZ: ROB-55.2-2532.Vet_02-15-176). A knockout of platelet sGC can be reached via deleting the sGC α1 or β1 subunit. Both
subunits form a functional unit so a knockout can be generated by knocking out either subunit. Since the knockout of sGC β1 has been established in cell-specific models42,43, we chose to
study sGC β1 knockout mice although the gene encoding sGC α1 was primarily identified as a CAD risk gene4,5. Platelet-specific sGC knockout mice (Pf4-_Cre__+__Gucy1b1__LoxP_/_LoxP_) were
obtained by crossing conditional NO-GC β1 knockout mice (_Gucy1b1__LoxP_/_LoxP_)44 and Pf4-_Cre__tg_/+ mice (Pf4-_Cre_+) as described previously45. Subsequently,
Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP_ mice were crossbred with _Ldlr__−/−_ mice (B6.129S7-_Ldlr_tm1Her/J) to induce an _Ldlr__−/−_ pro-atherosclerotic background. For experiments,
Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP__Ldlr_−/− and _Gucy1b1_+/_LoxP__Ldlr_−/− mice were mated to receive control (Pf4-_Cre_+_Gucy1b1_+/_LoxP__Ldlr_−/−) and platelet-specific sGC knockout
(Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP__Ldlr_−/−) littermates. _Irag1__LoxP_/_LoxP_ mice46 were mated with Pf4-_Cre_+ mice and obtained Pf4-Cre+_Irag1__LoxP_/_LoxP_ mice were backcrossed to a
C57BL/6J background. Pf4-_Cre_+_Irag1_+/_LoxP_ and Pf4-_Cre_+_Irag1_+/+ littermates were used as controls. _Ldlr__−/−_, C57BL/6J (WT) and _Ubc-GFP_ (C57BL/6-Tg(UBC-GFP)30Scha/J) mice were
purchased from The Jackson Laboratory. Only male or male and female mice at a 1:1 ratio at 8–12 weeks of age were used for all experiments. Mice were kept in a specific pathogen-free area
with HEPA-filtered room air. Cages were illuminated with an automatic light regime of 12 h in a day–night rhythm and temperature was kept constant between 20 and 22 °C at a humidity of
45–60%. To compare aortic plaque sizes between Pf4-_Cre__+__Gucy1b1__LoxP_/_LoxP__Ldlr_−/− and Pf4-_Cre_+_Gucy1b1_+/_LoxP__Ldlr_−/− mice, animals were fed a Western diet (21.2% fat and 0.2%
cholesterol by weight; TD.88137; Envigo) for 10 weeks. For intravital fluorescence microscopy, Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP__Ldlr_−/− and Pf4-_Cre_+_Gucy1b1_+/_LoxP__Ldlr_−/− mice were
fed a Western diet (21.2% fat and 0.2% cholesterol by weight; TD.88137) for 6 weeks. To compare aortic plaque sizes after pharmacological sGC stimulation, _Ldlr__−/−_ mice were fed a Western
diet (21.1% crude fat and 0.15% cholesterol, E15721-34; TD.88137 modified; ssniff, Soest) containing either 0 (control group) or 150 ppm (treatment group) of the sGC stimulator BAY-747
(N-(2-amino-2-methylbutyl)-8-((2,6-difluorobenzyl)oxy)-2,6-dimethylimidazo(1,2-a]pyridine-3-carboxamide) (Bayer AG) ad libitum for 10 weeks. For adoptive transfer experiments, _Ldlr__−/−_
mice were fed the same diet for six weeks. HUMAN SAMPLES The study protocol was approved by the local ethics committee of the Technical University of Munich (no. 387/17S). Blood was
collected from healthy volunteers after signing the informed consent. To determine the genotype of the individuals, DNA was isolated from whole blood using the Puregene Blood Kit (catalog
no. 158489; QIAGEN) according to the manufacturer’s protocol. Samples were genotyped for the _GUCY1A1_ risk variant using an rs7692387 TaqMan Genotyping Assay (C__29125113_10) on a Viia7
system (both Thermo Fisher Scientific). HISTOLOGY, IMMUNOHISTOCHEMISTRY AND EN FACE STAINING Aortic roots were embedded in optimal cutting temperature compound (catalog no. 62550; Sakura
Finetek) and snap-frozen to −80 °C. Frozen samples were cut into 5-µm sections and applied to microscope slides. From the onset of aortic valves, every fifth slide was subjected to tissue
staining. For the Masson’s trichrome stain, the procedure of Masson as modified by Lillie was applied according to the manufacturer’s instructions (Sigma-Aldrich). In brief, frozen sections
were hydrated and fixated in 4% paraformaldehyde before mordanting in Bouin’s solution (catalog no. HT10132; Sigma-Aldrich) at 56 °C for 15 min. Afterwards, cell nuclei were stained in
Weigert’s iron hematoxylin solution (catalog no. HT1079; Sigma-Aldrich) and darkened in running tap water. Specimens were successively subjected to Biebrich scarlet-acid fuchsin solution,
phosphotungstic/phosphomolybdic acid solution and aniline blue solution (catalog no. HT15; Sigma-Aldrich) for staining of cytoplasm, muscle and collagen structures, respectively. After
rinsing in 1% acetic acid, slides were dehydrated in an increasing ethanol row followed by xylene and covered with mounting medium. Mean total plaque size (in µm2) was evaluated for sections
showing at least two complete cusps by manually selecting the plaque area in ImageJ2 (version 2.3.0). For immunohistochemistry, specimens were fixed in ice-cold acetone, blocked in 10%
rabbit serum (catalog no. S5000; Vector Laboratories) in PBS with Tween-20 (0.2%) and stained in anti-CD11b antibody (1:200, catalog no. 101202; BioLegend) or anti-monocyte + macrophage
(MOMA) antibody (1:50, catalog no. ab33451; Abcam) overnight, followed by incubations in horseradish peroxidase (HRP)-conjugated rabbit anti-rat secondary antibody (1:200, catalog no.
ab6734; Abcam) and AEC substrate (catalog no. ab64252; Abcam). The primary antibodies have been validated by the manufacturers for use in immunohistochemistry on frozen mouse samples.
Subsequently, cell nuclei were counterstained with Gill’s hematoxylin solution II (catalog no. 1051752500; Merck Millipore). CD11b or monocyte and macrophage content was quantified as CD11b-
or MOMA2-positive area, respectively, per total plaque area by means of automated color thresholding in ImageJ2. For en face analyses, aortae were dissected from the heart to the iliac
bifurcation, cleaned of surrounding tissue and fixed for 24 h at 4 °C in a 4% solution of paraformaldehyde in PBS. Samples were washed in 60% isopropanol and incubated for 30 min at 37 °C in
a freshly filtered solution of 3 mg ml−1 Oil Red O (ORO) (catalog no. O0625; Sigma-Aldrich) in 60% isopropanol. After washing off excess dye in 60% isopropanol, aortae were opened
longitudinally, pinned on a black pad and imaged on a Stemi 2000-C microscope with an Axiocam ERc 5 s camera using the ZEN 2.3 blue software (version 2.3.69.1000, Carl Zeiss). The percentage
of the lesion area was determined manually as the ORO-positive area of total en face aortic area from aortic root to branch of the right renal artery using ImageJ2. All staining and
analyses were performed in blinded fashion. ISOLATION AND CULTURE OF BONE MARROW MEGAKARYOCYTES Bones of Pf4-_Cre_+_Gucy1b1_+/_LoxP_ and Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP_ mice were
centrifuged at 2,500_g_ for 1 min after removing proximal epiphyses47. The obtained bone marrow was subjected to 1× RBC lysis buffer (catalog no. 420301; BioLegend), filtered through 70-µm
mini-cell strainers and resuspended in IMDM medium (catalog no. 31980030; Thermo Fisher Scientific) supplemented with 10% FCS (catalog no. S0615; Sigma-Aldrich), penicillin-streptomycin
(1:100, catalog no. 15070063; Thermo Fisher Scientific), thrombopoietin (200 ng ml−1, catalog no. 130-096-301; Miltenyi Biotec) and stem cell factor (20 ng ml−1, catalog no. 130-101-693;
Miltenyi Biotec) to a concentration of 1 × 107 cells per ml. Cells were cultivated in a humidified incubator with 5% CO2 at 37 °C for 9 d and supplemented with fresh medium every third day.
Megakaryocytes were collected by performing two rounds of bovine serum albumin (BSA) density gradient filtration48. Briefly, cells were resuspended in PBS and placed on top of two layers of
a 1.5 and 3% BSA solution and incubated for 40 min. Sedimented cells were subjected to a second round of density gradient filtration, obtained purified megakaryocytes resuspended in 500 µl
of TRIzol (catalog no. 15596026; Thermo Fisher Scientific) and stored at −80 °C for further processing. ISOLATION OF NUCLEIC ACIDS AND (QUANTITATIVE) PCR After adding chloroform, samples
were shaken vigorously and centrifuged at 12,000_g_ for 15 min and 4 °C. The upper phase containing RNA was further processed using the RNeasy Mini Kit (catalog no. 74139; QIAGEN) according
to the manufacturer’s recommendations. RNA was quantified using a NanoQuant Plate on an Infinite M200 PRO plate reader (TECAN) and RNA integrity was measured on a 2100 Bioanalyzer (Agilent
Technologies). After the RNA was transcribed into complementary DNA using the High-Capacity RNA-to-cDNA kit (catalog no. 4388950; Applied Biosystems), real-time quantitative PCR (qPCR) was
performed using the TaqMan Fast Universal PCR Master Mix (catalog no. 4366072) and TaqMan probes (_Gucy1a1_, Mm01220285_m1; _Gucy1b1_, Mm00516926_m1; _Gucy1a2_, Mm01253540_m1; _Gucy1b2_,
Mm00555742_m1; _Vcam1_, Mm01320970_m1; _Gapdh_, Mm99999915_g1; all Thermo Fisher Scientific). Reactions were performed in a total volume of 10 µl on a ViiA 7 system (Thermo Fisher
Scientific). _Gapdh_ was used as a housekeeping gene and data were evaluated by conversion to ΔCt values. PROTEIN EXTRACTION Lung and aorta were collected from Pf4-_Cre_+_Gucy1b1_+/_LoxP_
and Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP_ mice after perfusion of organs with PBS and snap-frozen in liquid nitrogen. For protein isolation, specimens were placed in ice-cold
radioimmunoprecipitation assay (RIPA) buffer (catalog no. 9806S; Cell Signaling Technology) supplemented with 1:100 protease inhibitor cocktail (catalog no. 1861278; Thermo Fisher
Scientific) and disrupted using an electric tissue homogenizer on ice. To isolate peripheral blood mononuclear cells (PBMCs), heparinized full blood was applied onto a layer of Ficoll-Paque
Premium (catalog no. 17-5442-02; GE Healthcare) and centrifuged at 400_g_ for 30 min without break. The interface containing PBMCs was transferred to new tubes, washed and incubated in RBC
lysis buffer (BioLegend) for 5 min. Cells were resuspended in RIPA supplemented with 1:100 protease inhibitor cocktail. For the generation of thrombocyte lysates, platelets were collected
from heparinized full blood as stated previously and resuspended in RIPA buffer supplemented with protease inhibitor to a number of 2 × 108 cells per ml. Cells were disrupted by intermittent
sonication 3 times for 30 s in an ice bath. Protein concentrations were determined using a bicinchoninic acid assay (catalog no. 23227; Thermo Fisher Scientific) according to the
manufacturer’s protocol. IMMUNOBLOTTING Samples were supplemented with 4× Laemmli buffer (catalog no. 1610747; Bio-Rad Laboratories) containing 355 mM 2-mercaptoethanol and denatured for 5
min at 95 °C. Proteins were separated by SDS–gel electrophoresis using 4–20% Mini-PROTEAN TGX precast gradient gels (catalog no. 4561094; Bio-Rad Laboratories) at 100 V for 1.5 h in 1×
Tris/glycine/SDS buffer (catalog no. 161-0772; Bio-Rad Laboratories). Wet blotting (25 mM Tris, 192 mM glycine, 20% v/v methanol, pH 8.3) was performed at 100 V for 90 min using
methanol-activated polyvinylidene difluoride membranes. Membranes were blocked for 1 h at room temperature in PBS-T (PBS with 0.2% Tween) containing 5% nonfat dry milk powder (NFDM, catalog
no. A0830; AppliChem). Primary antibodies (anti-mβ1, directed against the β1 subunit of the sGC44 diluted 1:1,000 and GAPDH, catalog no. 8884S; Cell Signaling Technology, 1:200,000 in 2.5%
NFDM-PBS-T) were incubated at 4 °C overnight, followed by a 1-h incubation at room temperature with anti-rabbit HRP-conjugated secondary antibody (1:100,000 in 2.5% NFDM-PBS-T, catalog no.
7074; Cell Signaling Technology). For signal detection, membranes were developed with SuperSignal West Dura Extended Duration Substrate (catalog no. 34075; Thermo Fisher Scientific)
according to the manufacturer´s recommendations and signal intensities were detected using an ImageQuant 800 imager (GE Healthcare). PLATELET AGGREGATION Platelet-rich plasma (PRP) was
collected from Pf4-_Cre_+_Gucy1b1_+/_LoxP_ and Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP_ mice as stated in the manuscript and thrombocyte count was measured using an automated hematology analyzer
(XP-300; Sysmex Corp). PRP was centrifuged at 700_g_ for 10 min to receive PPP used for blanking. Samples were incubated at 37 °C in glass cuvettes with constant stirring on an 8-channel
personal computer-controlled platelet aggregation profiler (PAP-8, Biodata Corp) either in the presence of sodium nitroprusside (final concentration 10 µmol l−1, catalog no. HN34.1; Carl
Roth), BAY-747 (150 ppm) or vehicle (dimethyl sulfoxide (DMSO), 0.4%) for 2 min. Subsequently, thrombocyte aggregation was induced by the addition of ADP (final concentration 2 µmol l−1,
catalog no. 0203001; mölab), Ala-Tyr-Pro-Gly-Lys-Phe-NH2 (final concentration 75 µM, catalog no. A3227; Sigma-Aldrich), U46619 (final concentration 5 µM, catalog no. 1932; Tocris Bioscience)
and collagen (final concentration 1 µg ml−1, catalog no. 0203009). Platelet aggregation was recorded over 5 min to measure the area under the curve and displayed as a.u. min. GENERATION OF
SUPERNATANT FROM ACTIVATED PLATELETS A total of 800 µl each of blood from Pf4-_Cre_+_Gucy1b1_+/_LoxP_ mice and Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP_ mice was collected in heparinized tubes,
gently diluted in PBS and centrifuged for 10 min at 100_g_ and room temperature without active deceleration. PRP was subjected to a second centrifugation step at 700_g_ to obtain platelets
that were resuspended in Roswell Park Memorial Institute (RPMI) 1640 medium (catalog no. A1049101; Thermo Fisher Scientific) and activated by orbital shaking for 30 min at 1,000 rpm. Samples
were centrifuged at 12,000_g_ for 10 min and the supernatant was directly used in subsequent experiments. Thrombocyte count was analyzed simultaneously with an automated hematology analyzer
(Sysmex Corporation). For sGC stimulation, platelet suspension of WT mice was split in 2 vials containing either BAY-747 (150 ppm (150 mg l−1) final concentration) or vehicle (DMSO, 0.4%),
mixed gently and incubated for 30 min before shaking. Blood from healthy volunteers was collected in hirudin-coated tubes (Sarstedt) and centrifuged for 13 min at 170 g and room temperature
without active deceleration. PRP was transferred into new tubes and activated by orbital shaking for 30 min at 1,000 rpm. Afterwards, samples were centrifuged at 12,000 _g_ for 10 min and
the supernatant was stored at −80 °C before conducting subsequent experiments. Thrombocyte count was determined from PRP as stated above. IN VITRO LEUKOCYTE ADHESION Bone marrow monocytes
and neutrophils were isolated from C57BL/6J mice using magnetic-activated cell sorting cell separation columns (catalog no. 130-042-401; Miltenyi Biotec) after incubation with either
anti-Ly6G-PE (clone 1A8; catalog no. 127608) or anti-CD115-biotin (clone AFS98; catalog no. 135508, both BioLegend and 1:200) antibodies followed by phycoerythrin (PE)- and
streptavidin-coated microbeads (catalog nos. 130-048-801 and 130-048-101; Miltenyi Biotec), respectively. Primary murine aortic ECs (catalog no. C57-6052, mAoEC; Cell Biologics) were
cultured in complete EC medium (catalog no. PB-M1168; PeloBiotech) in a humidified incubator with 5% CO2 at 37 °C and grown to confluency in 96-well plates for experiments. Adhesion assays
were performed using the CytoSelect Leukocyte-Endothelium Adhesion Assay (catalog no. CBA-210; Cell Biolabs) according to the manufacturer’s instructions. Briefly, leukocytes were
fluorescently labeled with LeukoTracker solution, resuspended in RPMI 1640 and added to mAoECs to a number of 2.5 × 105 cells. Cells were incubated for 1 h at 37 °C in the presence of 50 µl
of activated platelet supernatant. Plates were washed three times to remove nonadherent cells, lysed in 1× lysis buffer and fluorescence was measured on an Infinite M200 PRO plate reader
(Exc = 485 nm and Em = 535 nm; TECAN). Experiments were conducted in triplicate. For the preincubation experiments, either neutrophils/monocytes or ECs were exclusively incubated with
activated platelet supernatant of WT mice for 30 min before performing the adhesion assay, omitting the additional administration of platelet supernatant in this step. To inhibit the Tie2
receptor effects, mAoECs were preincubated with 0.5 µM BAY-826 (catalog no. 6579; R&D Systems) or vehicle (DMSO, 0.1%) for 5 h before performing the adhesion assay. INCUBATION OF ECS
WITH ACTIVATED PLATELET SUPERNATANT mAoECs were grown to confluence in 12-well dishes and stimulated with activated platelet supernatant from either Pf4-_Cre_+_Gucy1b1_+/_LoxP_ or
Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP_ mice in RPMI 1640 for 1 h at 37 °C. Cells were lysed by adding 500 µl TRIzol and stored at −80 °C. INTRAVITAL FLUORESCENCE MICROSCOPY AND IN VIVO LEUKOCYTE
ADHESION Pf4-_Cre_+_Gucy1b1_+/_LoxP__Ldlr_−/− and Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP__Ldlr_−/− mice were anesthetized with a combination of midazolam, medetomidine and fentanyl and subjected to
intravital microscopy of the right carotid artery bifurcation as described previously49. Leukocytes were labeled in vivo by intravenous injection of anti-Ly6G (PE-conjugated, clone 1A8,
catalog no. 127608), anti-Ly6C (Alexa Fluor 488-conjugated, clone HK1.4, catalog no. 128022) and anti-CD11b (PE-conjugated, clone M1/70, catalog no. 101225; all BioLegend) antibodies (1 µg
each per mouse). To examine leukocyte to endothelium interactions, movies of 30 s each were acquired using an Olympus BX51 microscope with a Hamamatsu 9100-02 electron-multiplying
charge-coupled device camera (Hamamatsu) and a 10× saline-immersion objective. In subsequent analysis of the movies, the number of adherent neutrophils (Ly6G-PE+), monocytes (Ly6C-Alexa
Fluor 488+), and myeloid cells (CD11b-PE+) was examined in a blinded manner, considering cells as adherent if their position did not change during imaging. CYTOKINE PROFILING AND
ENZYME-LINKED IMMUNOSORBENT ASSAYS Cytokine profiling assays were performed using the Proteome Profiler Mouse XL Cytokine Array (catalo no. ARY028; R&D Systems) according to the
manufacturer’s protocol. Briefly, after isolating and activating the platelets of Pf4-_Cre_+_Gucy1b1_+/_LoxP_ and Pf4-_Cre_+_Gucy1b1__LoxP_/_LoxP_ mice by shaking in RPMI as described above,
samples were added to the supplied antibody-spotted nitrocellulose membrane and incubated at 4 °C overnight. Captured proteins were detected using a mixture of biotinylated detection
antibodies followed by streptavidin-HRP and visualized using chemiluminescent detection reagents. Signal intensities were detected by an ImageQuant LAS 4000 imaging system and analyzed using
the appropriate image analysis software (ImageQuant LAS TL, version 8.1; GE Healthcare Life Sciences). The signal intensities of target proteins were normalized to the signal intensities
for the reference spots in each sample. The procedure was repeated for a total of six samples per group. ANGPT1 ELISAs were performed to determine murine (catalog no. EK1296; Boster
Biological Technology) and human (catalog no. DANG10; R&D Systems) protein levels according to the manufacturer’s recommendations. COEXPRESSION ANALYSES IN STARNET Aligned multitissue
RNA-seq samples from STARNET15 were pseudo-log transformed and normalized using L2 penalized regression with penalty term 1.0, adjusting for the covariates: sequencing laboratory; read
length; RNA extraction protocol (PolyA and Ribo-Zero); age; and sex. Additional adjustments included the first four surrogate variables detected by surrogate variable analysis50 and flow
cell information after singular value decomposition retaining components with eigenvalues >4. Coexpression modules were inferred using weighted gene coexpression network analysis51 with
_β_ = 5.2 for tissue-specific and _β_ = 2.7 for cross-tissue correlations, resulting in both tissue-specific and cross-tissue coexpression networks as described previously52. These data and
analyses were accessed through the STARNET browser (http://starnet.mssm.edu) by querying coexpression modules containing ANGPT1. KEGG pathway and GO enrichment was carried out on
coexpression module 11 for transcripts derived from whole blood (1,012 out of 1,016 transcripts) using Enrichr53. FLOW CYTOMETRY Mice were killed under isoflurane anesthesia and blood was
collected in EDTA-coated microvettes (Sarstedt). For in vivo staining of circulating blood leukocytes, an antibody directed against CD45-BV605 (1:10 in 100 µl PBS, clone 30-F11, catalog no.
103140; BioLegend) was injected intravenously 5 min before killing the animals. After lysing RBCs in 1× RBC lysis buffer, samples were washed and resuspended in fluorescence-activated cell
sorting (FACS) buffer (PBS containing 0.5% BSA). Aortas were perfused through the left ventricle with PBS and excised from root to common iliac artery bifurcation after removing perivascular
fat and surrounding other tissue, minced using fine scissors and digested in 450 U ml−1 collagenase I (catalog no. C0130), 125 U ml−1 collagenase XI (catalog no. C7657), 60 U ml−1 DNase I
(catalog no. D5319) and 60 U ml−1 hyaluronidase (catalog no. H3506; all Sigma-Aldrich) for 1 h at 37 °C under agitation. Cell suspensions were filtered through 40-µm nylon cell strainers (BD
Biosciences), washed and resuspended in FACS buffer. Both blood and aortic cells were first stained for hematopoietic lineage markers with PE-conjugated antimouse antibodies directed
against B220 (1:600, clone RA3-6B2, catalog no. 103208), CD49b (1:1200, clone DX5, catalog no.108908), CD90.2 (1:3,000, clone 53-2.1, catalog no. 140308), NK1.1 (1:600, clone PK136, catalog
no. 108708), Ter119 (1:600, clone TER-119, catalog no. 116208), and Ly6G (1:600, clone 1A8, catalog no. 127608) for 15 min at 4 °C and washed. This was followed by a second round of staining
for Ly6C-BV421 (clone HK4.1, catalog no. 128031), CD115-BV510 (clone AFS98, catalog no. 750893; BD Biosciences), CD45.2-PerCP/Cy5.5 (1:300, clone 104, catalog no. 109827), F4/80-PE-Cy7
(clone BM8, catalog no. 123113), and CD11b-APC-Cy7 (clone M1/70, catalog no. 101225) in 1:600 dilution, if not stated otherwise. Antibodies were purchased from BioLegend if not stated
otherwise and validated by the manufacturers for use in flow cytometry. Cells were submitted to flow cytometry analysis on a BD LSRFortessa (BD Biosciences) and analyzed with the FlowJo
software v.9.9.6 (FlowJo LLC). Cells were gated on viable (forward scatter area (FSC-A) versus side scatter area (SSC-A)) and single (FSC-A versus forward scatter width (FSC-W) and SSC-A
versus side scatter width) cells. Neutrophils were identified as lineagehigh(CD45.2/CD11b)highCD115low. Monocytes were identified as lineagelow(CD45.2/CD11b/CD115)highLy6Chigh/low.
Macrophages were identified as lineagelow(CD45.2/CD11b/F4/80)highLy6Clow/int. PAMGENE SERINE/THREONINE KINASE ARRAY Washed platelets from WT mice were incubated with vehicle or BAY-747 (150
ppm) and activated by shaking as described above. Cells were centrifuged at 1,200_g_ for 5 min and resuspended in ice-cold M-PER lysis buffer (catalog no. 78503) supplemented with protease
inhibitor (1:100) and phosphatase inhibitor (1:100, catalog no. 78428; all Thermo Fisher Scientific) to a concentration of 200 × 106 platelets per 100 µl. Platelets were lysed for 15 min on
ice. Lysates were centrifuged at 20,000_g_ for 15 min and stored at −80 °C. Serine/threonine kinase profiles were determined using the PamChip serine/threonine kinase assay (PamGene
International) as described previously54. INHIBITION OF PLATELET SIGNALING PATHWAYS Washed platelets were isolated from WT mice as described above and incubated with different inhibitors of
downstream cGMP signaling (Supplementary Table 1) compared to vehicle for 30 min at room temperature. Subsequently, platelets were activated by shaking for 30 min, centrifuged, and
supernatant was collected for determination of ANGPT1 release as described above. DETERMINATION OF BAY-747 SERUM/PLASMA CONCENTRATION BAY-747 exposure was quantified in plasma using a liquid
chromatography (LC) system for mass separation (Kinetex 5 µm C18 100 A LC Column 150 × 4.6 mm) coupled to a Triple Quad 4500 LC–mass spectrometry analyzer (positive mode; AB Sciex). A
generic internal standard was added to the samples. A five-point calibration curve and quality control samples were used for relative quantification. Plasma was obtained from six mice per
group. Data are the mean + s.e.m. ADOPTIVE TRANSFER OF LEUKOCYTES Bone marrow monocytes and neutrophils were isolated simultaneously from _Ubc-GFP_ mice using anti-Ly6G-PE and
anti-CD115-biotin antibodies followed by PE- and streptavidin-coated microbeads as stated above and resuspended in PBS. We intravenously injected equal amounts of isolated cells into
_Ldlr_−/− mice fed a 0 or 150 ppm BAY-747 containing Western diet for 6 weeks and collected blood and aortae 24 h later as stated above. The number of CD45.2high/CD11bhigh/GFPhigh cells
within the aorta normalized to the exact number of injected cells was quantified by flow cytometry. STATISTICAL ANALYSIS Normality distribution of the data was assessed using the
Kolmogorov–Smirnov test or the Shapiro–Wilk test for sample sizes _n_ < 5. Test results and subsequently used statistical tests are displayed in Supplementary Table 2. Data were analyzed
using a two-tailed Student’s unpaired or paired _t_-test (for normally distributed data) or Mann–Whitney _U_-test (for non-normally distributed data), as appropriate and indicated in the
respective figure legend and Supplementary Table 2. When comparing more than two groups, a repeated measures one-way analysis of variance (ANOVA) test followed by a Tukey test for multiple
comparisons or mixed-effect analyses/repeated measures one-way ANOVA were performed, as appropriate, when data were normally distributed. To determine the statistical outliers, the two-sided
ROUT test was used. If outliers were removed from the analysis, this is indicated in the respective figure legend. Sample sizes/numbers of replicates are indicated in the figure legends and
visualized in the figures (each symbol represents one animal or biological replicate) and data are displayed as the mean + s.e.m. _P_ values <0.05, when investigating more than two
groups after adjustment for multiple testing, were regarded as statistically significant. Statistical analyses were performed with Prism v.9 for macOS (GraphPad Software). REPORTING SUMMARY
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY The data supporting the findings of this study are
available within the paper and its extended data/supplementary material. The STARNET data are available at the database of Genotypes and Phenotypes (dbGaP) site (dbGaP study accession no.
phs001203.v1.p1). REFERENCES * Benjamin, E. J. et al. Heart disease and stroke statistics–2019 update: a report from the American Heart Association. _Circulation_ 139, e56–e528 (2019).
Article Google Scholar * Erdmann, J., Kessler, T., Venegas, L. M. & Schunkert, H. A decade of genome-wide association studies for coronary artery disease: the challenges ahead.
_Cardiovasc. Res._ 114, 1241–1257 (2018). CAS Google Scholar * Dang, T. A., Schunkert, H. & Kessler, T. cGMP signaling in cardiovascular diseases: linking genotype and phenotype. _J.
Cardiovasc. Pharmacol._ 75, 516–525 (2020). Article CAS Google Scholar * Deloukas, P. et al. Large-scale association analysis identifies new risk loci for coronary artery disease. _Nat.
Genet._ 45, 25–33 (2013). Article CAS Google Scholar * Erdmann, J. et al. Dysfunctional nitric oxide signalling increases risk of myocardial infarction. _Nature_ 504, 432–436 (2013).
Article CAS Google Scholar * Wobst, J. et al. Stimulators of the soluble guanylyl cyclase: promising functional insights from rare coding atherosclerosis-related _GUCY1A3_ variants.
_Basic Res. Cardiol._ 111, 51 (2016). Article Google Scholar * Kessler, T. et al. Functional characterization of the _GUCY1A3_ coronary artery disease risk locus. _Circulation_ 136,
476–489 (2017). Article CAS Google Scholar * Emdin, C. A. et al. Phenotypic consequences of a genetic predisposition to enhanced nitric oxide signaling. _Circulation_ 137, 222–232 (2018).
Article CAS Google Scholar * Hall, K. T. et al. Genetic variation at the coronary artery disease risk locus _GUCY1A3_ modifies cardiovascular disease prevention effects of aspirin. _Eur.
Heart J._ 40, 3385–3392 (2019). Article Google Scholar * Kessler, T., Schunkert, H. & von Hundelshausen, P. Novel approaches to fine-tune therapeutic targeting of platelets in
atherosclerosis: a critical appraisal. _Thromb. Haemost._ 120, 1492–1504 (2020). Article Google Scholar * Kim, I., Moon, S. O., Park, S. K., Chae, S. W. & Koh, G. Y. Angiopoietin-1
reduces VEGF-stimulated leukocyte adhesion to endothelial cells by reducing ICAM-1, VCAM-1, and E-selectin expression. _Circ. Res._ 89, 477–479 (2001). Article CAS Google Scholar * Kim,
I. et al. Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3′-Kinase/Akt signal transduction pathway. _Circ. Res._ 86, 24–29 (2000). Article CAS Google
Scholar * Biswas, S. et al. IRAG1 deficient mice develop PKG1β dependent pulmonary hypertension. _Cells_ 9, 2280 (2020). Article CAS Google Scholar * Webb, T. R. et al. Systematic
evaluation of pleiotropy identifies 6 further loci associated with coronary artery disease. _J. Am. Coll. Cardiol._ 69, 823–836 (2017). Article CAS Google Scholar * Franzén, O. et al.
Cardiometabolic risk loci share downstream _cis_- and _trans_-gene regulation across tissues and diseases. _Science_ 353, 827–830 (2016). Article Google Scholar * Koplev, S. et al. A
mechanistic framework for cardiometabolic and coronary artery diseases. _Nat. Cardiovasc. Res._ 1, 85–100 (2022). Article Google Scholar * Zeng, L. et al. Contribution of gene regulatory
networks to heritability of coronary artery disease. _J. Am. Coll. Cardiol._ 73, 2946–2957 (2019). Article CAS Google Scholar * Ghofrani, H.-A. et al. Riociguat for the treatment of
pulmonary arterial hypertension. _N. Engl. J. Med._ 369, 330–340 (2013). Article CAS Google Scholar * Armstrong, P. W. et al. Vericiguat in patients with heart failure and reduced
ejection fraction. _N. Engl. J. Med._ 382, 1883–1893 (2020). Article CAS Google Scholar * Dubey, R. K., Jackson, E. K. & Luscher, T. F. Nitric oxide inhibits angiotensin II-induced
migration of rat aortic smooth muscle cell. Role of cyclic-nucleotides and angiotensin1 receptors. _J. Clin. Invest._ 96, 141–149 (1995). Article CAS Google Scholar * Mellion, B. T. et
al. Evidence for the inhibitory role of guanosine 3′, 5′-monophosphate in ADP-induced human platelet aggregation in the presence of nitric oxide and related vasodilators. _Blood_ 57, 946–955
(1981). Article CAS Google Scholar * Nikpay, M. et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. _Nat. Genet._ 47, 1121–1130
(2015). Article CAS Google Scholar * Nelson, C. P. et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. _Nat. Genet._ 49, 1385–1391
(2017). Article CAS Google Scholar * Dang, T. A. et al. Identification of a functional _PDE5A_ variant at the chromosome 4q27 coronary artery disease locus in an extended myocardial
infarction family. _Circulation_ 144, 662–665 (2021). Article Google Scholar * Wen, L. et al. A shear-dependent NO-cGMP-cGKI cascade in platelets acts as an auto-regulatory brake of
thrombosis. _Nat. Commun._ 9, 4301 (2018). Article Google Scholar * Nykänen, A. I. et al. Angiopoietin-1 protects against the development of cardiac allograft arteriosclerosis.
_Circulation_ 107, 1308–1314 (2003). Article Google Scholar * Fujisawa, T. et al. Angiopoietin-1 promotes atherosclerosis by increasing the proportion of circulating Gr1+ monocytes.
_Cardiovasc. Res._ 113, 81–89 (2017). Article CAS Google Scholar * Gamble, J. R. et al. Angiopoietin-1 is an antipermeability and anti-inflammatory agent in vitro and targets cell
junctions. _Circ. Res._ 87, 603–607 (2000). Article CAS Google Scholar * Antl, M. et al. IRAG mediates NO/cGMP-dependent inhibition of platelet aggregation and thrombus formation. _Blood_
109, 552–559 (2007). Article CAS Google Scholar * Sandner, P. et al. Soluble guanylate cyclase stimulators and activators. _Handb. Exp. Pharmacol._ 264, 355–394 (2021). Article CAS
Google Scholar * Ahluwalia, A. et al. Antiinflammatory activity of soluble guanylate cyclase: cGMP-dependent down-regulation of P-selectin expression and leukocyte recruitment. _Proc. Natl
Acad. Sci. USA_ 101, 1386–1391 (2004). Article CAS Google Scholar * Tsou, C.-Y. et al. Activation of soluble guanylyl cyclase prevents foam cell formation and atherosclerosis. _Acta
Physiol. (Oxf.)_ 210, 799–810 (2014). Article CAS Google Scholar * Klarin, D. et al. Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to
coronary artery disease. _Nat. Genet._ 49, 1392–1397 (2017). Article CAS Google Scholar * Melichar, V. O. et al. Reduced cGMP signaling associated with neointimal proliferation and
vascular dysfunction in late-stage atherosclerosis. _Proc. Natl Acad. Sci. USA_ 101, 16671–16676 (2004). Article CAS Google Scholar * Andersson, D. P. et al. Association of
phosphodiesterase-5 inhibitors versus alprostadil with survival in men with coronary artery disease. _J. Am. Coll. Cardiol._ 77, 1535–1550 (2021). Article CAS Google Scholar * Bennett, B.
J. et al. Genetic architecture of atherosclerosis in mice: a systems genetics analysis of common inbred strains. _PLoS Genet._ 11, e1005711 (2015). Article Google Scholar * Maynard, D.
M., Heijnen, H. F. G., Horne, M. K., White, J. G. & Gahl, W. A. Proteomic analysis of platelet α-granules using mass spectrometry. _J. Thromb. Haemost._ 5, 1945–1955 (2007). Article CAS
Google Scholar * Chatterjee, M. et al. Distinct platelet packaging, release, and surface expression of proangiogenic and antiangiogenic factors on different platelet stimuli. _Blood_ 117,
3907–3911 (2011). Article CAS Google Scholar * Italiano, J. E. Jr et al. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate
platelet α granules and differentially released. _Blood_ 111, 1227–1233 (2008). Article CAS Google Scholar * Sehgal, S. & Storrie, B. Evidence that differential packaging of the major
platelet granule proteins von Willebrand factor and fibrinogen can support their differential release. _J. Thromb. Haemost._ 5, 2009–2016 (2007). Article CAS Google Scholar * Neagoe,
P.-E., Brkovic, A., Hajjar, F. & Sirois, M. G. Expression and release of angiopoietin-1 from human neutrophils: intracellular mechanisms. _Growth Factors_ 27, 335–344 (2009). Article
CAS Google Scholar * Groneberg, D. et al. Smooth muscle-specific deletion of nitric oxide-sensitive guanylyl cyclase is sufficient to induce hypertension in mice. _Circulation_ 121,
401–409 (2010). Article CAS Google Scholar * Groneberg, D. et al. Cell-specific deletion of nitric oxide-sensitive guanylyl cyclase reveals a dual pathway for nitrergic neuromuscular
transmission in the murine fundus. _Gastroenterology_ 145, 188–196 (2013). Article CAS Google Scholar * Friebe, A., Mergia, E., Dangel, O., Lange, A. & Koesling, D. Fatal
gastrointestinal obstruction and hypertension in mice lacking nitric oxide-sensitive guanylyl cyclase. _Proc. Natl Acad. Sci. USA_ 104, 7699–7704 (2007). Article CAS Google Scholar *
Rukoyatkina, N., Walter, U., Friebe, A. & Gambaryan, S. Differentiation of cGMP-dependent and -independent nitric oxide effects on platelet apoptosis and reactive oxygen species
production using platelets lacking soluble guanylyl cyclase. _Thromb. Haemost._ 106, 922–933 (2011). Article CAS Google Scholar * Desch, M. et al. IRAG determines nitric oxide- and atrial
natriuretic peptide-mediated smooth muscle relaxation. _Cardiovasc. Res._ 86, 496–505 (2010). Article CAS Google Scholar * Heib, T., Gross, C., Müller, M.-L., Stegner, D. & Pleines,
I. Isolation of murine bone marrow by centrifugation or flushing for the analysis of hematopoietic cells—a comparative study. _Platelets_ 32, 601–607 (2021). Article CAS Google Scholar *
Drachman, J. G., Sabath, D. F., Fox, N. E. & Kaushansky, K. Thrombopoietin signal transduction in purified murine megakaryocytes. _Blood_ 89, 483–492 (1997). Article CAS Google Scholar
* Winter, C. et al. Chrono-pharmacological targeting of the CCL2–CCR2 axis ameliorates atherosclerosis. _Cell Metab._ 28, 175–182.e5 (2018). Article CAS Google Scholar * Leek, J. T.,
Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. _Bioinformatics_ 28,
882–883 (2012). Article CAS Google Scholar * Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. _BMC Bioinformatics_ 9, 559 (2008). Article
Google Scholar * Talukdar, H. A. et al. Cross-tissue regulatory gene networks in coronary artery disease. _Cell Syst._ 2, 196–208 (2016). Article CAS Google Scholar * Kuleshov, M. V. et
al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. _Nucleic Acids Res._ 44, W90–W97 (2016). Article CAS Google Scholar * Peters, L. J. F. et al.
MicroRNA-26b attenuates platelet adhesion and aggregation in mice. _Biomedicines_ 10, 983 (2022). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank C. Wolf for
technical assistance. This work is funded by the Corona Foundation as part of the Junior Research Group Translational Cardiovascular Genomics (no. S199/10070/2017 to T.K.) and the German
Research Foundation as part of the collaborative research centers SFB 1123 (A6 to O.S. and B02 to T.K. and H.S.), SFB 1009 (A13 to O.S.), TRR 267 (B06 to H.S.), TRR 332 (A02/Z01 to O.S.),
KE2116/1-4 (to T.K.) and STRESS_638675 (to H.B.S.). Further grants were received from the European Research Council under the European Union’s Horizon 2020 research and innovation program
(grant no. 759272 to H.B.S.) and the German Heart Foundation (F/28/17 to H.B.S.) as well as the German Federal Ministry of Education and Research within the framework of ERA-NET on
Cardiovascular Disease (grant no. JTC2017_21-040 to H.S. and J.L.M.B.) and within the scheme of target validation (BlockCAD: 6GW0198K to H.S.). Additional support was received from the
British Heart Foundation/German Centre for Cardiovascular Research collaborative project ‘Genetic discovery-based targeting of the vascular interface in atherosclerosis’, the Leducq
Foundation (PlaqOmics; clonal hematopoiesis and atherosclerosis to J.L.M.B.), the Swedish Research Council (no. 2018-02529_VR to J.L.M.B.), the Swedish Heart-Lung-Foundation (no. 20200207 to
J.L.M.B.), the Interdisciplinary Center for Clinical Research within the faculty of Medicine at the Research Rhine-Westphalia Technical University of Aachen and the Fritz Thyssen Foundation
(grant no. 10.20.2.043MN, both to E.P.C.v.d.V.). AUTHOR INFORMATION Author notes * These authors contributed equally: Carina Mauersberger, Hendrik B. Sager. * These authors jointly
supervised this work: Heribert Schunkert, Thorsten Kessler. AUTHORS AND AFFILIATIONS * German Heart Centre Munich, Department of Cardiology, Technical University of Munich, Munich, Germany
Carina Mauersberger, Hendrik B. Sager, Jana Wobst, Tan An Dang, Laura Lambrecht, Marlène Stroth, Noomen Bettaga, Heribert Schunkert & Thorsten Kessler * German Centre for Cardiovascular
Research, Munich Heart Alliance, Munich, Germany Carina Mauersberger, Hendrik B. Sager, Jana Wobst, Tan An Dang, Laura Lambrecht, Marlène Stroth, Oliver Soehnlein, Heribert Schunkert &
Thorsten Kessler * Department of Genetics and Genomic Sciences, Icahn Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, NY, USA Simon Koplev
& Johan L. M. Björkegren * Cancer Research UK Cambridge Institute, University of Cambridge, Li Ka Shing Centre, Cambridge, UK Simon Koplev * Department of Pharmacology and Toxicology,
University of Regensburg, Regensburg, Germany Jens Schlossmann * Bayer AG, R&D Pharmaceuticals, Wuppertal, Germany Frank Wunder, Lisa Dietz & Peter Sandner * Institute of Physiology,
Julius Maximilian University of Würzburg, Würzburg, Germany Andreas Friebe * Department of Medicine, Neo, Karolinska Institutet, Karolinska Universitetssjukhuset, Huddinge, Sweden Johan L.
M. Björkegren * Department of Cardiac Surgery and The Heart Clinic, Tartu University Hospital and Department of Cardiology, Institute of Clinical Medicine, Tartu University, Tartu, Estonia
Johan L. M. Björkegren * Institute for Molecular Cardiovascular Research and Interdisciplinary Centre for Clinical Research, Rhine-Westphalia Technical University of Aachen, Aachen, Germany
Sanne L. Maas & Emiel P. C. van der Vorst * Institute for Cardiovascular Prevention, Ludwig Maximilian University of Munich, Munich, Germany Emiel P. C. van der Vorst & Oliver
Soehnlein * Institute for Experimental Pathology, University of Münster, Münster, Germany Oliver Soehnlein * Department of Physiology and Pharmacology and Department of Medicine, Karolinska
Institutet, Stockholm, Sweden Oliver Soehnlein Authors * Carina Mauersberger View author publications You can also search for this author inPubMed Google Scholar * Hendrik B. Sager View
author publications You can also search for this author inPubMed Google Scholar * Jana Wobst View author publications You can also search for this author inPubMed Google Scholar * Tan An
Dang View author publications You can also search for this author inPubMed Google Scholar * Laura Lambrecht View author publications You can also search for this author inPubMed Google
Scholar * Simon Koplev View author publications You can also search for this author inPubMed Google Scholar * Marlène Stroth View author publications You can also search for this author
inPubMed Google Scholar * Noomen Bettaga View author publications You can also search for this author inPubMed Google Scholar * Jens Schlossmann View author publications You can also search
for this author inPubMed Google Scholar * Frank Wunder View author publications You can also search for this author inPubMed Google Scholar * Andreas Friebe View author publications You can
also search for this author inPubMed Google Scholar * Johan L. M. Björkegren View author publications You can also search for this author inPubMed Google Scholar * Lisa Dietz View author
publications You can also search for this author inPubMed Google Scholar * Sanne L. Maas View author publications You can also search for this author inPubMed Google Scholar * Emiel P. C.
van der Vorst View author publications You can also search for this author inPubMed Google Scholar * Peter Sandner View author publications You can also search for this author inPubMed
Google Scholar * Oliver Soehnlein View author publications You can also search for this author inPubMed Google Scholar * Heribert Schunkert View author publications You can also search for
this author inPubMed Google Scholar * Thorsten Kessler View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS H.B.S., H.S. and T.K. generated the
hypothesis and conceived the project. C.M., H.B.S., O.S., H.S. and T.K. designed the experiments. C.M., H.B.S., J.W., T.A.D., L.L., M.S., N.B., L.D., P.S. and O.S. performed the experiments.
C.M., H.B.S., J.W., S.K., L.D., P.S., J.L.M.B., O.S. and T.K. analyzed and interpreted the data. F.W., L.D., A.F., J.S., S.L.M, E.P.C.v.d.V., P.S. and J.L.M.B. provided data and/or
materials. All authors provided intellectual input and edited the manuscript. C.M., H.B.S., H.S. and T.K. drafted the manuscript, which was edited and approved by all authors. CORRESPONDING
AUTHORS Correspondence to Hendrik B. Sager, Heribert Schunkert or Thorsten Kessler. ETHICS DECLARATIONS COMPETING INTERESTS H.S. has received personal fees from MSD Sharp & Dohme, Amgen,
Bayer Vital GmbH, Boehringer Ingelheim, Daiichi Sankyo, Novartis, Servier, Brahms, Bristol Myers Squibb, Medtronic, Sanofi Aventis, Synlab, Pfizer and Vifor T as well as grants and personal
fees from AstraZeneca that are unrelated to the submitted work. H.S. and T.K. are named inventors on a patent application for the prevention of restenosis after angioplasty and stent
implantation (patent applicants: Klinikum rechts der Isar, German Heart Centre Munich; inventors: T. Kessler, A. Kastrati, H. Schunkert; application no. PCT/EP2021/053116; status: pending),
which is unrelated to the submitted work. T.K. received lecture fees from Bayer HealthCare Pharmaceuticals. L.D., F.W. and P.S. are full-time employees of Bayer HealthCare Pharmaceuticals.
The other authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Cardiovascular Research_ thanks Gillian Douglas and the other, anonymous, reviewer(s) for their
contribution to the peer review of this work. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional
affiliations. EXTENDED DATA EXTENDED DATA FIG. 1 SGC-EXPRESSION IN PF4-_CRE_+_GUCY1B1__LOXP/LOXP_ COMPARED TO PF4-_CRE_+_GUCY1B1_+/_LOXP_ MICE. A. Expression of sGC-β1 in platelets,
peripheral blood mononuclear cells (PBMC), aorta, and lung. Representative of three independently performed Western blots on different samples. B+C. _Gucy_ transcript expression analysis in
megakaryocytes of Pf4-_Cre_+_Gucy1b1_+/_LoxP_ and Pf4-_Cre_+_Gucy1b1__LoxP/LoxP_ mice. B. _Gucy1a1_ and _Gucy1b1_. C. _Gucy1a2_ and _Gucy1b2_. Each symbol represents one independent animal
(n = 6 for control group, n = 5 for Pf4-_Cre_+_Gucy1b1__LoxP/LoxP_). Two-sided unpaired t-test. Data are mean and s.e.m. Source data EXTENDED DATA FIG. 2 PLATELET AGGREGATION IN CONTROL AND
PLATELET SGC KNOCKOUT (PF4-_CRE_+_GUCY1B1__LOXP/LOXP_) MICE. Platelet aggregation in control and platelet sGC knockout (Pf4-_Cre_+_Gucy1b1__LoxP/LoxP_) mice after stimulation with adenosine
diphosphate (ADP, A) the platelet-activated receptor 4 agonist PAR4-AP (B), the thromboxane analog U46619 (C), and collagen (D). Each experiment was performed in the presence of DMSO
(vehicle) or the nitric oxide donor sodium nitroprusside (SNP). Each symbol represents one independent animal (n = 6). Aggregation tracings represent the mean values of investigated animals
per genotype. Two-sided unpaired t-test (except A, vehicle and D, vehicle: two-sided Mann-Whitney U-test). Data are mean and s.e.m. Source data EXTENDED DATA FIG. 3
PF4-_CRE_+_GUCY1B1__LOXP/LOXP__LDLR_−/− COMPARED TO PF4-_CRE_+_GUCY1B1_+/_LOXP__LDLR_−/− MICE AFTER WESTERN DIET (SEE FIG. 1E). A. Serum cholesterol levels (n = 11). B. Platelet count (n =
11). C. Body weight (n = 11) at baseline (left) and after ten weeks (right). D. Blood leukocyte numbers and subsets (n = 11 for controls, n = 10 for Pf4-_Cre_+_Gucy1b1__LoxP/LoxP__Ldlr_−/−).
Each symbol represents one independent animal. Two-sided unpaired t-test (except C 10 weeks: two-sided Mann-Whitney U-test). One outlier was removed in the analysis of D according to the
ROUT method. Data are mean and s.e.m. Source data EXTENDED DATA FIG. 4 LEUKOCYTE ADHESION TO ATHEROSCLEROTIC PLAQUES OF PF4-_CRE_+_GUCY1B1__LOXP/LOXP__LDLR_−/− (N = 13) COMPARED TO
PF4-_CRE_+_GUCY1B1_+/_LOXP__LDLR_−/− MICE (N = 11) AFTER WESTERN DIET ASSESSED BY FLUORESCENCE INTRAVITAL MICROSCOPY. A. Ly6Chigh monocytes. B. Neutrophils. Each symbol represents one
independent animal. Two-sided unpaired t-test. Data are mean and s.e.m. Source data EXTENDED DATA FIG. 5 QUANTIFICATION OF MONOCYTE ADHESION AFTER PREINCUBATION OF EITHER EC OR MONOCYTES
WITH SUPERNATANT OF ACTIVATED PLATELETS FROM PF4-_CRE_ + _GUCY1B1__LOXP/LOXP_ MICE. Each symbol represents one paired sample (derived from n = 8 independent animals). Two-sided paired
t-test. Data are mean and s.e.m. Abbreviations: _EC_, endothelial cells; _rfu_, relative fluorescence units. Source data EXTENDED DATA FIG. 6 _VCAM1_ EXPRESSION OF WILD-TYPE ENDOTHELIAL
CELLS AFTER INCUBATION WITH SUPERNATANT OF ACTIVATED PF4-_CRE_+_GUCY1B1_+/_LOXP_ OR PF4-_CRE_+_GUCY1B1__LOXP/LOXP_ PLATELETS. Each symbol represents one independent animal (n = 6). Data are
mean and s.e.m. Two-sided paired t-test. Source data EXTENDED DATA FIG. 7 CYTOKINE PROFILING. A-H. Cytokine profiling assay results for cytokines detected at a mean relative intensity ≥1 (in
addition to Angiopoietin-1). I. Replication attempt for RBP4 release using ELISA. Each symbol represents one independent animal (A-H: n = 8; I: n = 5). Two-sided unpaired t-test (except B
and G: Two-sided Mann-Whitney U-test). Data are mean and s.e.m. Abbreviations: _au_, arbitrary units; _CXCL5_, C-X-C Motif Chemokine Ligand 5; _IGFPB2_, Insulin Like Growth Factor Binding
Protein 2; _RBP4_, Retinol Binding Protein 4; _REG3G_, Regenerating islet-derived protein 3 gamma. Source data EXTENDED DATA FIG. 8 ANGIOPOIETIN-1 (ANGPT1) RELEASE BY CONTROL OR
PF4-_CRE_+_IRAG1__LOXP/LOXP_ PLATELETS AFTER ACTIVATION BY SHAKING. Each symbol represents one independent animal (n = 6). Two-sided unpaired t-test. Data are mean and s.e.m. Source data
EXTENDED DATA FIG. 9 ADENOSINE DIPHOSPHATE-INDUCED PLATELET AGGREGATION IN PF4-_CRE_+_GUCY1B1_+/_LOXP_ AND PF4-_CRE_+_GUCY1B1__LOXP/LOXP_ PLATELETS AFTER PRETREATMENT WITH VEHICLE OR
BAY-747. Each symbol represents one independent animal (n = 3). Two-sided unpaired t-test. Data are mean and s.e.m. Abbreviation: _ADP_, adenosine diphosphate. Source data EXTENDED DATA FIG.
10 CHARACTERISTICS OF _LDLR_−/− MICE RECEIVING 0 PPM COMPARED TO _LDLR_−/− MICE RECEIVING 150 PPM BAY-747 AFTER TEN WEEKS OF WESTERN DIET. A. Serum cholesterol levels (n = 12). B. Platelet
count (n = 9 in 0 ppm, n = 8 in 150 ppm group). C. Blood leukocytes (n = 9 in 0 ppm, n = 8 in 150 ppm group). D. Body weight after (n = 12). E. Plasma concentrations of the soluble guanylyl
cyclase stimulator BAY-747 in treated mice. F. Blood leukocyte numbers and subsets. Each symbol represents one independent animal (n = 12). Two-sided unpaired t-test. Two outliers were
removed in the analysis of F: neutrophils (150 ppm group; n = 10) and Ly6Chigh monocytes (150 ppm group; n = 11) according to the ROUT method. Data are mean and s.e.m. Source data
SUPPLEMENTARY INFORMATION REPORTING SUMMARY SUPPLEMENTARY TABLES 1–6. SOURCE DATA SOURCE DATA FIG. 1 Statistical source data. SOURCE DATA FIG. 2 Statistical source data. SOURCE DATA FIG. 3
Statistical source data. SOURCE DATA FIG. 4 Statistical source data. SOURCE DATA FIG. 5 Statistical source data. SOURCE DATA EXTENDED DATA FIG. 1 Statistical source data. SOURCE DATA
EXTENDED DATA FIG. 1 Uncropped western blot (Extended Data Fig. 1a). SOURCE DATA EXTENDED DATA FIG. 2 Statistical source data. SOURCE DATA EXTENDED DATA FIG. 3 Statistical source data.
SOURCE DATA EXTENDED DATA FIG. 4 Statistical source data. SOURCE DATA EXTENDED DATA FIG. 5 Statistical source data. SOURCE DATA EXTENDED DATA FIG. 6 Statistical source data. SOURCE DATA
EXTENDED DATA FIG. 7 Statistical source data. SOURCE DATA EXTENDED DATA FIG. 8 Statistical source data. SOURCE DATA EXTENDED DATA FIG. 9 Statistical source data. SOURCE DATA EXTENDED DATA
FIG. 10 Statistical source data. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons
license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a
credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted
use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT
THIS ARTICLE CITE THIS ARTICLE Mauersberger, C., Sager, H.B., Wobst, J. _et al._ Loss of soluble guanylyl cyclase in platelets contributes to atherosclerotic plaque formation and vascular
inflammation. _Nat Cardiovasc Res_ 1, 1174–1186 (2022). https://doi.org/10.1038/s44161-022-00175-w Download citation * Received: 27 January 2022 * Accepted: 27 October 2022 * Published: 12
December 2022 * Issue Date: December 2022 * DOI: https://doi.org/10.1038/s44161-022-00175-w SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get
shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative