Sm-omics is an automated platform for high-throughput spatial multi-omics
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT The spatial organization of cells and molecules plays a key role in tissue function in homeostasis and disease. Spatial transcriptomics has recently emerged as a key technique to
capture and positionally barcode RNAs directly in tissues. Here, we advance the application of spatial transcriptomics at scale, by presenting Spatial Multi-Omics (SM-Omics) as a fully
automated, high-throughput all-sequencing based platform for combined and spatially resolved transcriptomics and antibody-based protein measurements. SM-Omics uses DNA-barcoded antibodies,
immunofluorescence or a combination thereof, to scale and combine spatial transcriptomics and spatial antibody-based multiplex protein detection. SM-Omics allows processing of up to 64 in
situ spatial reactions or up to 96 sequencing-ready libraries, of high complexity, in a ~2 days process. We demonstrate SM-Omics in the mouse brain, spleen and colorectal cancer model,
showing its broad utility as a high-throughput platform for spatial multi-omics. SIMILAR CONTENT BEING VIEWED BY OTHERS THE EMERGING LANDSCAPE OF SPATIAL PROFILING TECHNOLOGIES Article 20
July 2022 REPURPOSING LARGE-FORMAT MICROARRAYS FOR SCALABLE SPATIAL TRANSCRIPTOMICS Article 19 November 2024 SPATIAL MULTI-OMICS AT SUBCELLULAR RESOLUTION VIA HIGH-THROUGHPUT IN SITU
PAIRWISE SEQUENCING Article 14 May 2024 INTRODUCTION The spatial organization of cells and molecules is fundamental to physiological function and disease pathology, and imaging the position
and level of molecules is a cornerstone of both basic biology and clinical pathology. Because gene expression is regulated at multiple levels from transcription to protein degradation,
protein and RNA levels convey distinct information on gene function and cell state, as has been shown in diverse contexts including dynamic responses1,2, in genetic variation3, in human
malignancies4, and in single cells in suspension5. Single cell genomics and multi-omics approaches, such as single cell and single nucleus RNA-Seq6,7,8,9,10,11 and CITE-Seq5,12, have been
tremendously successful at capturing diverse molecular profiles at the level of individual cells and nuclei, but typically do not preserve spatial information. The importance of studying
cells in their native environment has been shown in many processes, from normal organ development to spatial deregulation in diseases and often highlighted in the context of cancer
propagation and resistance to therapy13,14. Recent progress in spatial in situ profiling methods has opened the way for comprehensive profiling of location and expression
simultaneously15,16,17,18,19,20,21,22,23,24,25,26,27,28. For spatial RNA measurements, Spatial Transcriptomics (ST)24,26 has emerged as a versatile approach for spatial RNA profiling. In ST,
a fresh-frozen tissue section is placed on top of barcoded DNA primers attached to a glass surface24. Following tissue staining and histological imaging, cells are permeabilized, mRNAs are
spatially tagged directly in tissues and a cDNA sequencing library is generated. After sequencing, the RNA-Seq information is traced back to the spatially barcoded positions on the glass
slide providing a global spatial tissue profile. ST has been applied to diverse systems and tissue types, such as brain, heart, spinal cord, melanomas, breast cancer and prostate
cancer24,29,30,31,32,33,34,35,36. However, barriers around throughput, resolution, and efficiency37, limit its application at large scale. In parallel, there have been advances in multiplex
protein measurements in situ based on reading out multiple fluorescent-, heavy metal- or barcode coupled antibody tags19,20,38,39,40,41. Some methods rely on cyclic immunostaining or in situ
sequencing barcoding schemes, whereas others use expensive machinery for Multiplexed Ion Beam Imaging or Imaging Mass Cytometry. Few platforms have combined RNA and antibody-based
measurements to date42,43,44 and have traditionally relied on imaging one or the other modality. Companion technologies similar to our approach (e.g. Visium, 10X Genomics) rely on: (i) an
antibody-based immunofluorescence (IF) read-out of 1–2 target antigens; (ii) do not employ DNA-barcoding strategies which allow us to parallelize antibody-based measurements, (iii) and
process spatial RNA-Seq libraries manually, making these approaches low-throughput, laborious and not scalable due to intrinsic limitations of multiplex imaging. To bridge this gap and make
molecular tissue profiling a widely available and robust tool, we develop Spatial Multi-Omics (SM-Omics), an end-to-end framework that uses a liquid handling platform for high-throughput
combined transcriptome and antibody-based spatial tissue profiling with minimum user input and available laboratory instrumentation45,46. SM-Omics relies on using DNA-barcoded antibodies,
similarly to how CITE-seq5 performs simultaneous epitope and transcriptome profiling in single cells, to scale and combine spatial transcriptomics and spatial antibody-based multiplex
protein detection. This user-friendly all-sequencing based technology allows processing of up to 64 in situ spatial reactions and up to 96 sequencing-ready libraries, of high complexity, in
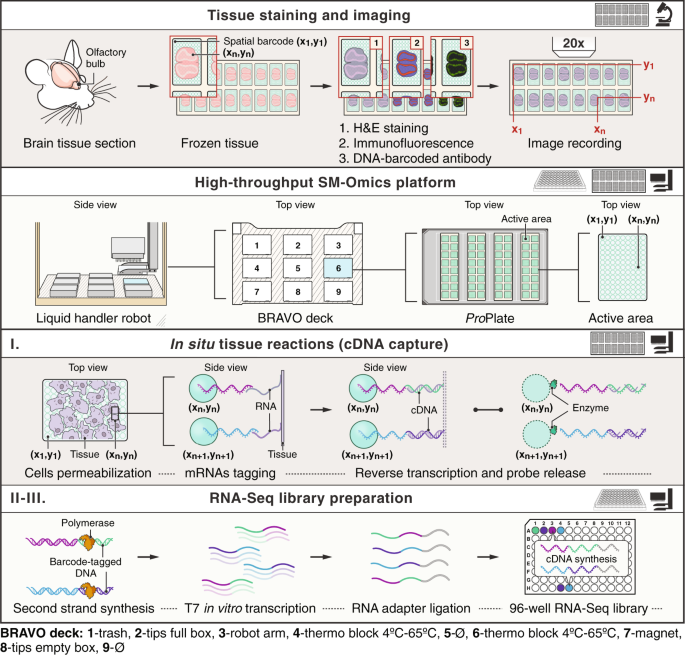
~2 days, in a high-throughput platform for spatial multi-omics. RESULTS We developed the SM-Omics platform for either automated Spatial Transcriptomics alone, or, in combination with
fluorescently or DNA-barcoded antibodies to simultaneously measure spatial profiles of RNAs and proteins. Briefly, in SM-Omics, after tissue staining for traditional hematoxylin and eosin
histology (H&E), IF or using DNA-barcoded antibodies, glass slides are loaded into the SM-Omics platform, where, using a liquid handler robot, cells are permeabilized, mRNAs and/or
antibody barcodes are spatially tagged and converted into a sequencing-ready library (Fig. 1). The automated process consists of three main parts with designed stopping points to either
store the processed material or load required reagents for the upcoming reactions. The first step consists of all in situ enzymatic reactions on the SM-Omics slide, including tissue
permeabilization after staining and reverse transcription with simultaneous release of spatial capture probes (Fig. 1I). Each such in situ run holds up to 4 slides with tissues, with the
number of active areas with spatial probes per slide ranging from one to 16 per slide. The second and third steps consist of RNA-Seq library preparation in standard 96 well plates, where the
user can choose to run between 1 and 96 libraries in parallel in 8-step increments with adjusted library consumable usage to alleviate costs. The input to these is in situ spatial tissue
cDNA or DNA-barcoded antibody tags captured from glass slides in the first step, which are then processed to amplify cDNA using a T7 in vitro transcription approach (for cDNA) or standard
PCR amplification (for DNA-barcoded antibody tags), followed by a final conversion of the amplified RNAs into sequencing-ready libraries (Fig. 1II, III). SM-Omics introduces four key
enhancements compared to ST: (1) automation, requiring minimal user intervention; (2) throughput, allowing processing of 96 libraries in a 2-day cycle; (3) enhanced quality, reflected by
higher complexity RNA-Seq libraries and (4) combining RNA-Seq measurements with multiplex protein measurements including IF staining and antibody-barcoding strategies. To process the
generated data efficiently, we also developed SpoTteR, a fast and fully automated end-to-end image registration method (Methods). We first describe the core approach in the context of
spatial RNA measurements (Fig. 1, II, III), and then its extension to include spatial antibody-based protein measurements. IN SITU SM-OMICS RNA-SEQ To test the performance of SM-Omics for
spatial transcriptomics, we assessed the feasibility, reproducibility and efficiency of RNA data in two key steps, testing on the mouse main olfactory bulb (MOB) and mouse cortex: (1) in
situ tissue reactions (cDNA capture) and (2) cDNA library preparation for RNA-Seq. SM-Omics had enhanced performance in terms of in situ reactions compared to standard ST, with minimal
lateral diffusion and comparable and reproducible cDNA signal intensity. Specifically, we first ran in situ reactions on the glass surface in optimization mode, where cDNA molecules are in
situ fluorescently labeled to create a spatial cDNA footprint36 (Supplementary Fig. 1a). We compared the localized cDNA footprint to the histological H&E pattern and measured the lateral
tissue permeabilization effects. This provides an optimal set of parameters needed to successfully run tissue-specific reactions and to ensure minimal lateral cross-talk between adjacent
spatial measurements. Testing on the adult mouse cortex (Supplementary Fig. 1b–e') showed that SM-Omics resulted in no mixing of material between spatial measurements with no lateral
diffusion (mean −0.06 µm ± 0.51 µm sd), which is 4x weaker lateral diffusion signal than in ST performed on adjacent tissue sections (two-sided Wilcoxon’s rank-sum test, _p_-value ≤ 0.05,
Supplementary Fig. 1f, g), and 30x weaker diffusion signal compared to previous reports24,36,47. Moreover, the signal intensity of the fluorescent cDNA footprint was highly reproducible
within and between SM-Omics runs: there were no significant differences (two-sided Wilcoxon’s rank-sum test, _p_-value > 0.05) between the cDNA signal intensities from adjacent adult
mouse MOB tissue replicates on a single glass slide (_n_ = 3), single run (_n_ = 3) or separate runs (_n_ = 3) (Supplementary Fig. 2). To process the generated data efficiently, we also
developed SpoTteR, a fast and fully automated end-to-end image registration method. SpoTteR automatically downscales images and reconstructs barcode spots positions using iterative spot
detection and grid fitting (Methods), accounting for common imaging artifacts, such as uneven tissue coloration or pipetting bubbles. SpoTteR then registers tissue coordinates through a
masking process to produce a gene-by-barcode matrix overlaid on top of morphological features (Supplementary Fig. 3). Compared to manual and semi-automated image registration approaches48,
SpoTter is up to 14X faster with low false discovery rates (FP 3.54% and FN 1.18%, _vs_. >15% of grid spots as FNs in other approaches48), when applied to images of human lung cancer,
human arthritis and mouse colon data (Supplementary Fig. 3b, Supplementary Fig. 4). Using the SM-Omics end-to-end toolbox (Fig. 1) we prepared and sequenced SM-Omics (_n_ = 3) high quality
RNA-Seq libraries (Supplementary Data 1) from the MOB of the adult mouse brain, and compared them to standard ST (_n_ = 3) libraries at the same sequencing depth (by down-sampling). SM-Omics
RNA-Seq libraries were more sensitive than ST, with a 3.2-fold higher number of unique protein-coding genes and a 3.6-fold higher number of unique transcripts (UMIs) present in the data
(Wald’s test, _p_-value ≤ 0.05, Methods, Supplementary Fig. 5a). Per spatial measurement, SM-Omics detected 2.5-fold more unique genes (3748 ± 562) and 3.5-fold more unique transcripts
(11,261 ± 2273 UMIs) than ST (1485 ± 185 genes; 3188 ± 513 UMIs) (Wald’s test, _p_ ≤ 0.05, Methods, Fig. 2a, Supplementary Fig. 5b). SM-Omics exhibited an increase on average (_n_ = 3) in
the number of transcripts captured in most of the annotated morphological regions compared to ST (Wald’s test, _p_-value ≤ 0.05, Methods, Supplementary Fig. 5c, Supplementary Data 2) and
performed comparably to newer array designs (_n_ = 3) (Visium, 10x Genomics, effect size = 2.11 and 1.38, for genes and UMIs respectively, _p_-value > 0.05, Wald’s test) (Fig. 2b,
Supplementary Fig. 5d, e). This increased efficiency in SM-Omics, as reflected in the number of genes and UMIs detected per (x, y) coordinate, was due to several optimizations in library
preparations. First, we introduced simultaneous release of barcoded primers and captured mRNA molecules (Methods) from the glass surface which also decreased total in situ processing time
from ~1.5 days to ~6 h. Second, we improved the efficiency of library preparation reactions, by increasing the amount of sequencing adapters and reaction time for adapter ligation to the
template (Methods, two-sided Wilcoxon’s rank-sum test, _p_ ≤ 0.05) (Fig. 2c). To further test SM-Omics RNA-Seq on challenging tissues, we optimized in situ reaction conditions for mouse
colon and colorectal cancer models, and found strong spatial fluorescent patterns in these tissues (Supplementary Fig. 6a, b). Moreover, SM-Omics (_n_ = 3) outperformed ST (_n_ = 3) in
library metrics per spatial measurement in the cancer model. SM-Omics detected significantly higher (Wald’s test, _p_-value ≤ 0.05, Supplementary Fig. 6c) numbers of genes and UMIs (5086 ±
121 genes and 16,250 ± 922 UMIs) compared to ST (2733 ± 492 genes and 5128 ± 1304 UMIs). We also compared SM-Omics and ST in terms of detecting spatial expression patterns. We used
Splotch32,49 to align an expanded dataset of 18 replicate MOB tissue sections and generate posterior spatial gene expression estimates. We confirmed that region-enriched and upregulated
genes were present in the major spatial layers (Methods) of the MOB compared to the reference Allen Brain Atlas (ABA) data50 (Supplementary Fig. 7a, b). While known gene patterns detected as
layer-enriched agreed between SM-Omics and ST (Supplementary Fig. 7c–f), SM-Omics’ overall specificity was higher (Supplementary Fig. 7a) and gene expression values per region were more
highly correlated between SM-Omics and ABA (Spearman’s ρ = 0.90, _p_-value ≤ 0.0001, Supplementary Fig. 7g) than between ST and ABA (Spearman’s ρ = 0.71, _p_-value ≤ 0.005, Supplementary
Fig. 7g). Compared to Visium, SM-Omics exhibited comparable regional metrics, with both methods showing enrichment of regionally expressed genes in the appropriate spatial layers of the
mouse brain cortex, and high correlation to expression levels in the ABA (Supplementary Fig. 8a, b), with similar regional sensitivity for both SM-Omics and Visium (Supplementary Fig. 8c).
This increased sensitivity (_vs_. ST) at the same sequencing depth (by down-sampling, Methods), allowed us to reproducibly measure the spatial gene expression of newly detected targets,
otherwise not detected by standard ST, such as _CTGF_ in the glomerular layer (GL) and _CAMK4_ in the granular cell layer (GR), both implicated in impairments in retention of long-term
memory51 and acting as targets of protein aggregation in models of Alzheimer’s disease52, as well as _LANCL3_ in the mitral layer (MI), _NR2F2_ in the olfactory nerve layer (ONL) and _CBLN4_
in the outer plexiform layer (OPL) (Fig. 2d, e). Identifying and quantifying these additional genes using SM-Omics’ increased sensitivity should help discover novel biological targets as
well as pursue hypothesis-driven research. SPATIAL TRANSCRIPTOMICS WITH ANTIBODY-BASED IMMUNOFLUORESCENCE We next developed a protocol that combined antibody-based IF with spatial
transcriptomics (Fig. 3a, Methods). Localized cDNA footprints after nuclear (DAPI) and IF staining of the tissue (Fig. 3b, Supplementary Fig. 9a) showed that mRNAs were laterally diffusing
only 0.16 ± 1.21 μm outside of the nucleus, again indicating minimal lateral cross-talk between adjacent spatial measurements. We next created SM-Omics mouse brain cortex libraries following
immunostaining with an antibody against the brain protein NeuN, which is highly expressed in most neuron nuclei (Fig. 3c). Library complexities, signal specificity and RNA expression
patterns were similar to those in standard (H&E stained) SM-Omics RNA-Seq measurements and in ABA50 (Supplementary Fig. 9b–d), confirming that our protocol for simultaneous IF and
transcriptome measurements provided high-quality mRNA data. Next, comparing the antibody IF signals and corresponding RNA expression (Fig. 3c), there was significant correlation between NeuN
mRNA and aggregated protein expression (Spearman’s ρ = 0.69, _n_ = 5, _p_-value ≤ 0.0001, Fig. 3c) across all major regions in the mouse brain cortex. Notably, in some regions (e.g.,
hypothalamus) RNA expression was low but protein expression was substantial (Fig. 3d). This may be due to either a biological difference, or to the differences in sensitivity and saturation
of RNA-Seq _vs_. IF. Furthermore, while throughput in antibody-based IF is limited and imaging data and mRNA data have different noise characteristics (Supplementary Data 2), it provides a
fast alternative to traditional H&E staining as well as adds quantitative protein information at single-cell resolution to any spatial array design. AN ALL-SEQUENCING-BASED APPROACH FOR
SPATIAL MULTI-OMICS Finally, we introduced an antibody DNA-barcoding system5 compatible with spatial transcriptomics to increase multiplexing capacities otherwise limited with spectral
overlap in imaging approaches (Fig. 4a). We tagged each of 6 antibodies5 with an amplification primer and an individual barcode tag followed by a poly(d)A sequence for capture on a poly(d)T
spatially barcoded array (Methods). We used a similar tissue staining protocol as that for IF, where the tissue was first in situ fixed with paraformaldehyde to ensure specific antigen
coupling, followed by antibody staining, tissue permeabilization and SM-Omics library preparations (Fig. 4a). To benchmark our approach, we incubated adult mouse spleen tissue sections with
both a fluorescently labeled antibody and a DNA-barcoded antibody (i.e. antibody tag), allowing us to simultaneously validate and directly compare both detection methods. We imaged the
fluorescently labeled epitopes prior to any in situ enzymatic reactions on the array surface, coupled the antibody tags to the spatial array, such that they were copied into a stable
covalent complex, while mRNA was spatially captured and transcribed on the array (Fig. 4a). We first tested a two-antibody cocktail targeting F4/80 and IgD (Fig. 4b), aimed to stain distinct
spatial niches in the mouse spleen: splenic red pulp macrophages and marginal zone B cells in the white pulp, as previously described19. We obtained high quality antibody tag (mean ± sd 142
± 15 UMIs per SM-Omics measurement; _n_ = 7) and cDNA libraries (1375 ± 181 UMIs per SM-Omics measurement, _n_ = 3), with highly specific antibody tag patterns (Fig. 4b) that were
well-correlated to the corresponding IF intensities across all major splenic regions (Supplementary Fig. 10a, on average 76%, _p_ ≤ 0.0001). RNA and antibody tag levels were in agreement for
IgD (Spearman’s ρ = 0.73, _n_ = 3, _p_-value ≤ 0.05 across all spatial measurements), and less so for F4/80 (Spearman’s ρ = 0.65, _n_ = 3, _p_-value ≤ 0.05 across all spatial measurements)
(Supplementary Fig. 10b). Finally, an SM-Omics experiment with six validated53 DNA-barcoded antibodies targeting F4/80, IgD, Cd163, Cd38, Cd4, and Cd8a (Supplementary Fig. 10c), spanning
different levels of expression and spatial patterns, successfully combined spatial transcriptomics and protein estimates in a highly multiplexed manner (Fig. 4c, Supplementary Fig. 10d). Cd4
and Cd8 proteins (by antibody signal) and their corresponding mRNAs were spatially localized in the PALS zone (Spearman’s ρ = 0.59, _n_ = 3, _p_-value ≤ 0.05), whereas IgD and Cd38 protein
and mRNA were enriched in the B follicles (Spearman’s ρ = 0.66, _n_ = 3, _p_-value ≤ 0.05), with protein expression high in all white pulp areas (Supplementary Fig. 10d). F4/80 protein and
mRNA were localized to the red pulp, but the corresponding mRNA (_ADGRE1_) was also enriched in the marginal zone (Supplementary Fig. 10d). Finally, Cd163 was differentially expressed, as
expected, in the red pulp, however, _CD163_ mRNA was high, apart from the red pulp zonations in PALS as well, while protein levels were not detected at significant levels in that same tissue
area (_p_-value > 0.05, Supplementary Fig. 10d). DISCUSSION SM-Omics is an efficient and automated workflow for combined and spatially resolved transcriptomics and antibody-based protein
measurements, adaptable to new array versions and designs. SM-Omics provides a more detailed molecular high-plex multi-omics characterization of tissues in situ and is a high-throughput
automated system for quantifying the spatial transcriptome and antibody-based protein detection, by either IF or using DNA-barcoded antibodies. Compared to approaches with similar array
design versions (Visium, 10X Genomics), SM-Omics provides an automated workflow that is not limited to performing a small number of high-resolution spatial IF measurements but further
extends the combined spatial transcriptomics and spatial antibody-based protein measurements into a scalable all-sequencing based technology. Using a 6-plex proof-of-concept antibody
SM-Omics reaction, we confirmed that SM-Omics is a robust system that can reconstruct specific cell associations across morphological layers54,55, and characterize tissue niches in
combination with antibody staining, which provide higher resolution views independently of or in combination with spatial transcriptomics patterns. SM-Omics can be enhanced in the future in
several ways, including demonstrating higher multiplex for protein detection (similarly to CITE-seq5), automating tissue sectioning workflows, increasing the resolution of spatial
measurements (to achieve that of recent spatial RNA-seq approaches26,56,57,58) and furthering work on integrating robust image registration and IF pipelines to aid in interpreting combined
signals from different modalities created with SM-Omics. Moreover, SM-Omics is currently limited to frozen tissues (whereas many clinical samples are FFPE) and to lower resolution arrays,
and future studies can tackle those to extend its applicability and enhance its resolution, respectively. Finally, while current costs of commercial spatial arrays might be limiting
(Supplementary Data 1), high throughput processing should motivate economies of scale. SM-Omics automation on a widely-used platform enables use of appropriate study design while minimizing
technical variation, and allowing broad adoption. Additionally, even if only used as a spatial transcriptomics library preparation system, its 96-plex throughput outperforms previous
automated protocol designs by 6–8 fold34,35. SM-Omics does not rely on any customized liquid handling microfabrication, uses commercially, widely-available liquid handlers and reagents with
minimum preparation time per run (~30 min), has an end-to-end image-integrated data registration pipeline and is readily deployable to the wide scientific community. METHODS ETHICAL
STATEMENT All work involving C57BL/6 J mice was performed under specific-pathogen-free conditions and the guidelines of the Division of Comparative Medicine, in accordance with the
Institutional Animal Care and Use Committees (IACUC) relevant guidelines at the Broad Institute of Harvard and MIT, and consistent with the Guide for Care and Use of Laboratory Animals,
National Research Council, 1996 (institutional animal welfare assurance no. A4711-01), with protocol 0122-10-16. BRAVO SYSTEM REQUIREMENTS Bravo Automated Liquid Handling Platform (Agilent
Technologies, USA) was equipped with a 96LT pipetting head (G5498B#042, Agilent Technologies, USA) and two Peltier thermal stations (CPAC Ultraflat HT 2-TEC, #7000166 A, Agilent
Technologies, USA) with PCR adapter having a mounting frame at positions 4 and 6 on the Bravo Deck and connected to an Inheco MTC Controller. On position 7, we recommend the MAGNUM FLX™
Enhanced Universal Magnet Plate (#A000400, Alpaqua, USA) to serve for magnetic bead-based clean ups. In addition, a BenchCel NGS Workstation (Front-load rack at 660 mm height) and BenchCel
Configuration Labware MiniHub (option #010, Agilent Technologies, USA) were included in the automation platform setup. In case in situ reactions were performed, the PCR adapter was removed
from position 6 to be replaced with Aluminum Heat Transfer Plate (#741I6-GS-4, V&P Scientific, Inc, USA). This liquid handling setup enables running in situ reactions using the ProPlate
Multi-Array slide system (GraceBioLabs, USA), where 64 reactions can be run in parallel using the standard 96LT pipetting head. Note that every third column in the 96-tip pipette box needs
to be removed when using the ProPlate Multi-Array system with standard Agilent Bravo pipetting instrumentation. All library preparation reactions are run in a maximum 96-well mode, however
lower throughput adjustments are predefined as 8-sample increments and easily loaded in our automated SM-Omics settings. Further details in the SM-Omics protocol sections below, and at:
https://github.com/klarman-cell-observatory/sm-omics/tree/master/SM_Omics_v.B1.0.2. SAMPLE COLLECTION AND CRYOSECTIONING All work involving C57BL/6 J mice was performed under
specific-pathogen-free conditions and the guidelines of the Division of Comparative Medicine, in accordance with the Institutional Animal Care and Use Committees (IACUC) relevant guidelines
at the Broad Institute of Harvard and MIT, and consistent with the Guide for Care and Use of Laboratory Animals, National Research Council, 1996 (institutional animal welfare assurance no.
A4711-01), with protocol 0122-10-16. A small piece of freshly collected tissue (~25–50 mg, about 5 × 5 mm) was placed on a dry and sterile Petri dish, which was placed on top of wet ice. The
tissue was then very gently moved using forceps and placed on another dry part of the Petri dish to ensure little liquid was present around the tissue. The bottom of a cryomold (5 × 5 mm,
10 × 10 mm or 25 × 20 mm) was filled with pre-chilled (4 °C) OCT (Tissue-Tek; Sakura Finetek, USA) and the tissue transferred with forceps into the OCT-prefilled mold. The entire tissue
surface was covered with pre-chilled OCT. The mold was then placed on top of dry ice and allowed the tissue to freeze for up to 5 min until OCT has turned completely white and hard. The
tissue cryomolds were stored at −80 °C until use. For cryosectioning, the ST slide and the tissue molds first reached the temperature of the cryo chamber. The OCT-embedded tissue block was
attached onto a chuck with pre-chilled OCT and allowed to freeze ~5–10 min. The chuck was placed in the specimen holder and adjusted the position to enable perpendicular sectioning at 10 µm
thickness. Sections were gently transferred to a ST array24 and then the back side of the slide was warmed ~10–15 s with a finger. ST slides with tissue sections on top could be stored at
−80 °C for up to 6 days. TISSUE FIXATION AND H&E STAINING The ST slide with the tissue section was warmed to 37 °C for 1 min on a thermal incubator (Eppendorf Thermomixer Option C,
Germany). The tissue was then covered with 4% formaldehyde (Sigma-Aldrich, USA) in 1X PBS (Thermo Fisher Scientific, USA) for 10 min at room temperature (RT). The whole slide was then washed
in 1X PBS in a vertical orientation to be placed back on a horizontal place for drying. 500 µl isopropanol covered the tissue and ensured drying. The slide was put into an EasyDip Slide Jar
Staining System (Weber Scientific) holder and the same system used for H&E staining. Five ~80 ml containers were prepared with Dako Mayers hematoxylin (Agilent, USA), Dako Bluing buffer
(Agilent, USA), 5% Eosin Y (Sigma–Aldrich, USA) in 0.45 M Tris acetate (Sigma–Aldrich, USA) buffer at pH 6 and two jars with nuclease-free water (ThermoFisher Scientific, USA). The slide
rack was fully immersed in hematoxylin for 6 min and then washed by dipping the slide rack in a nuclease-free water jar 5 times following another destaining wash by dipping the slide rack in
800 mL nuclease-free water for 30 times. The slide rack was put into the Dako bluing buffer and incubated for 1 min. The slide was again washed by dipping the rack 5 times in the second
nuclease-free water jar. The slide rack was finally put into the eosin and incubated for 1 min to be washed by dipping the rack 7 times in the second water jar. The slide was removed from
the rack to allow it to dry. TISSUE FIXATION AND IF STAINING The ST slide with the tissue section was warmed to 37 °C for 4 min on a thermal incubator (Eppendorf Thermomixer Option C,
Germany) and in situ fixed and washed as described above. The slide was then mounted in the plastic slide holder (ProPlate Multi-Array slide system; GraceBioLabs, USA) compatible with the
Aluminum Heat Transfer Plate (#741I6-GS-4, V&P Scientific, Inc, USA) on position 6 on the Bravo deck. All following antibody incubations were performed at 4 °C. First, the tissues were
blocked with the TruStain FcX™ PLUS (anti-mouse CD16/32, Biolegend, USA) antibody (1:100 dilution) in 0.5% Triton X-100 (Sigma-Aldrich, USA) for mouse brain tissues and 1× perm/wash buffer
(ThermoFisher Scientific, USA) for splenic tissues. This simultaneous blocking and permeabilization step lasted for 30 min. Next, the slide was washed 3× with 1× PBS (ThermoFisher
Scientific, USA). After discarding the last wash, the slides were incubated with 1× PBS for 2 min. Then, antibodies were added at 1:100 dilution for 90 min. The complete list of antibody
clones and suppliers is available in Supplementary Data 3. The slide was again washed in the same fashion and counterstained with DAPI (Sigma–Aldrich, USA) diluted 1:1000 in 0.5% Triton
X-100 (Sigma–Aldrich, USA) for 5 min. In case the reactions were performed on a SM-Omics array and not a mock polyd(T) array, the DAPI reaction was also supplemented with a Cy3 labeled
anti-frame DNA probe (5′-Cy3-GGTACAGAAGCGCGATAGCAG-3′, IDT, USA) at 10 nM concentration. In case DAPI counterstaining was not used, the step was skipped. This was followed by another wash
cycle. The slides were then air dried and mounted with 85% glycerol prior to imaging. TISSUE FIXATION AND DAPI-ONLY STAINING Similarly to performing _Tissue fixation and IF staining_, tissue
sections were attached to slides and in situ fixed. The slide was then mounted in the plastic slide holder (ProPlate Multi-Array slide system; GraceBioLabs, USA) and all reactions performed
at 4 °C. Tissues were first incubated with 0.5% Triton X-100 (Sigma–Aldrich, USA) for 25 min. Next, the slide was washed 1x PBS (ThermoFisher Scientific, USA) and the tissue stained with
DAPI (Sigma–Aldrich, USA) diluted 1:1000 in 0.5% Triton X-100 (Sigma–Aldrich, USA) for 15 min. If the reactions were performed on a SM-Omics array and not a mock polyd(T) array, the DAPI
reaction was also supplemented with a Cy3 labeled anti-frame DNA probe (5′-Cy3-GGTACAGAAGCGCGATAGCAG-3′, IDT, USA) at 10 nM concentration in order to facilitate image registration to the
SM-Omics array coordinates. This was followed by another wash cycle. The slides were then air dried and mounted with 85% glycerol prior to imaging. AUTOMATED IMAGING Images of stained
H&E tissue sections on the ST slides were taken using a Metafer Vslide scanning system (MetaSystems, Germany) installed on an Axio Imager Z2 microscope (Carl Zeiss, Germany) using an LED
transmitted light source and a CCD camera (BF scanning). All images were taken with the A-P 10x/0.25 Ph1 objective lens (Carl Zeiss, Germany). For fluorescent scanning, a PhotoFLuor LM-75
lightsource (89North, USA) was used in combination with a Plan-APOCHROMAT 20x/0.8 objective (Carl Zeiss, Germany). A configuration program was made to enable automatic tissue detection,
focusing and scanning on all ST arrays present on a glass slide. In short, tissue detection was based on contrast as compared to normalized background in all channels. Upon finding maximum
contrast in a 12-step spiral-like search window field of view (FOV) pattern, the automated focal alignment in every second of each FOV (4096 × 3000 px) was initiated. The alignment search
considered the maximum contrast z-position as in-focus using 5 µm stage intervals (_n_ = 19 focal planes). The BF scanning of the predefined ST array areas was done in a total of 48 FOVs and
~30 s in 3 channels (RGB); or epifluorescent scanning of 228 FOVs and ~6 min for 3 fluorescent channels. Images were stitched using 60 µm overlap and linear blending between FOVs with the
VSlide software (v1.0.0) and then extracted using jpg compression. Multiple ST slides can be processed in the same manner without any user input for a total of 6 min processing time per
H&E stained slide (3 channels) or 45 min for fluorescently stained slide (3 channels), including image stitching. MICROARRAY DESIGN AND PRODUCTION Both for quality control experiments
and library preparation, the Codelink amine activated slides (#DN01-0025, Surmodics, USA) were exposed with polyadenylated oligonucleotides (IDT, USA) and microarray production proceeded as
according to manufacturer’s instructions (Surmodics, USA). The surface oligonucleotides are presented here for clarity:
([AmC6]UUUUUGACTCGTAATACGACTCACTATAGGGACACGACGCTCTTCCGATCT[18nt]NNNNNNN[20 T]VN). This chemistry design enabled covalent linking upon binding to the Codelink slide surface. For library
preparation slide production, 33 μM spatially barcoded oligonucleotides (IDT, USA) were deposited as 100pL droplets onto Codelink slides as suggested by the manufacturer (Surmodics, USA).
This resulted in about ~200 million copies of the oligonucleotide per spatial spot. Array printing was performed by ArrayJet LTD (Scotland, UK) according to the ArrayJet Spider system
requirements. Each library preparation slide active area had a total of 1,007 spatially barcoded positions distributed over a ~42 mm2 area. Each spatially barcoded ST spot had a diameter of
100 μm, with a center-to-center distance of 200 μm between the spots. SM-OMICS AUTOMATION The SM-Omics protocol is divided into three main parts. The first part (1) processes all in situ
reactions on a ST slide: tissue pre-permeabilization, permeabilization, reverse transcription with or without the release of the spatial capture probes and tissue removal. This material is
collected to a standard 96-well PCR microplate (Eppendorf, Germany) and all of the following reactions (protocols 2 and 3) are run in 96-well plates. The second protocol (2) contains second
strand synthesis reaction, cDNA bead purifications and T7 in vitro transcription. The third protocol (3) includes aRNA adapter ligation, bead purifications and second cDNA synthesis. The
material is then quantified using a standard qPCR protocol and the libraries accordingly indexed for Illumina sequencing. REFERENCE MATERIAL PREPARATION In order to test reproducibility of
library preparation reactions, we prepared reference material as input. 7.5 µg of universal mouse reference RNA (#740100, Agilent Technologies, USA) was fragmented using NEBNext Magnesium
RNA fragmentation module (NEB, USA) for 1 min at 94 °C. The sample was purified with a MinElute Cleanup kit (Qiagen, Germany) according to manufacturer’s instructions and RNA concentration
and size were assessed using a Qubit RNA HS kit (ThermoFisher Scientific, USA) and Bioanalyzer Pico 6000 kit (Agilent Technologies, USA), respectively. ~2 µg of fragmented RNA was incubated
with either 3.3 µM custom hexamer primer (GACTCGTAATACGACTCACTATAGGGACACGACGCTCTTCCGATCTNNNNNNNN, T7handle_IlluminaAhandle_hexamer) or poly(d)T primer
(T7handle_IlluminaAhandle_hexamer_20TVN) in the presence of 0.8 mM dNTP (ThermoFisher Scientific, USA) at 65 °C for 5 min. First strand reverse transcription was performed with a final
concentration of 1X First Strand Buffer, 5 mM DTT, 2U/µl RNaseOUT and 20U/µl of Superscript III (all from Thermo Fisher Scientific, USA). The reaction was incubated at 25 °C for 10 min (when
using hexamer priming), followed by 50 °C for 1 h and 70 °C for 15 min or 50 °C for 1 h and 70 °C for 15 min for poly(d)T priming. The reaction was purified with AMPure XP beads (Beckman
Coulter, USA) at a beads/DNA ratio of 0.8:1. The concentration of the material was measured on a Qubit RNA HS kit (ThermoFisher Scientific, USA) and diluted in EB (Qiagen, Germany). A
release mixture of ~100 ng (hexamer priming) or ~200 ng (poly(d)T priming) first strand cDNA, 1X Second strand buffer (ThermoFisher Scientific, USA), 0.2 µg/µl BSA and 0.5 mM dNTP
(ThermoFisher Scientific, USA) was used to test all library preparation reactions. Hexamer primed cDNA was used to test the reproducibility and poly(d)T primed cDNA was used to test adapter
concentrations and ligation time. IN SITU SM-OMICS PROTOCOL (1) Tissue-stained ST slides we provided as input. The ST slide was attached into the ProPlate Multi-Array slide system
(GraceBioLabs, USA), with up to four ST slides fitted. The ProPlate Multi-Array system was then fixed in position by Aluminum Heat Transfer Plate (VP 741I6-GS-4, V&P Scientific, Inc,
USA) on the Agilent Bravo deck. The protocol started with tissue pre-permeabilization (30 min at 33 °C) with addition of 120 µl reagent per well of exonuclease I buffer for brain samples
(NEB, USA) or 120 µl reagent per well of collagenase I (200U) in 1x HBSS (both from Thermo Fisher Scientific, USA) for colorectal samples. For spleen sections, the pre-permeabilization step
was skipped. For complete removal of the reagents and wash solutions from the subarrays all of the robotic dispensing and aspiration steps took place in all four corners of the square wells.
Pre-permeabilization reagent removal was followed by a 180 µl wash in 0.1X Saline Sodium Citrate (SSC, Sigma–Aldrich, USA) at 33 °C. Next, tissue permeabilization was done using 75 µl 0.1%
pepsin (pH 1, Sigma–Aldrich, USA) at 33°C for 10 min (mouse brain) and 15 min (colorectal cancer) and for 60 min (spleen) 75 µl 0.1% pepsin prepared at pH 2.5 in Tris-HCl (Sigma-Aldrich,
USA). After a 180 µl 0.1X SSC wash at 33 °C, in situ cDNA synthesis reaction was performed by the addition of 75 µl RT reagents: 50 ng/µl actinomycin D (Sigma–Aldrich, USA), 0.5 mM dNTPs
(Thermo Fisher Scientific, USA), 0.20 µg/µl BSA, 1 U/µl USER enzyme (both from NEB, USA), 6% v/v lymphoprep (STEMCELL Technologies, Canada), 1 M betaine (#B0300-1VL, Sigma-Aldrich, USA), 1X
First strand buffer, 5 mM DTT, 2 U/µl RNaseOUT, 20 U/µl Superscript III (all from Thermo Fisher Scientific, USA). The reactions were sealed with 70 µl of white mineral oil Drakerol#7
(Penreco, USA). Incubation at 30 °C was performed for a minimum of 6 h, after which 70 µl of the released material was collected in a new 96-well PCR plate (Eppendorf, Germany). When a Cy3
fluorescent cDNA activity print was needed for tissue optimization, the 75 µl in situ cDNA reaction mix was as follows: 50 ng/µl actinomycin D (Sigma-Aldrich, USA), 0.20 µg/µl BSA (NEB,
USA), 1X M-MuLV buffer, 5 mM DTT, 2U/µl RNaseOUT, 20U/µl M-MuLV (all from Thermo Fisher Scientific, USA), 4 µl dNTP mix (dATP; dGTP and dTTP at 10 mM and dCTP at 2.5 mM) and 2.2 µl Cy3-dCTPs
(0.2 mM, Perkin Elmer, USA). IN SITU MANUAL ST PROTOCOL The manual ST in situ protocol was performed as described in Salmén et al.47. The protocol is, if not mentioned below, identical to
the robotic protocol except as further described. Tissue-stained ST slide was attached in an ArrayIT hybridization chamber (ArrayIT, CA). All incubations took place on an Eppendorf
Thermocycler R (Eppendorf, Germany), and reactions were covered with Microseal ‘B’ PCR Plate Seals (Biorad, CA) to avoid evaporation. Pre-permeabilization and washes were performed with 100
µl reagent at 37 °C and the in situ cDNA synthesis reaction was run without the USER enzyme, lymphoprep and betaine, at 42 °C. The manual protocol then encompassed tissue removal and probe
release as described47. Tissue removal took place in two separate steps with RLT buffer with β-mercaptoethanol and Proteinase K. 80 µl of 1% β-mercaptoethanol (Sigma-Aldrich, USA) in RLT
buffer (Qiagen, Germany) were added to the wells and incubated at 56°C for 1 h. Following removal of the reaction mix and wash with 0.1X SSC solution, 80 µl of second tissue removal mixture;
2.5 µg/µl Proteinase K in PDK buffer (Qiagen, Germany) were added and the reaction was performed at 56 °C for 1 h. The complete reaction mix was again removed and a slide wash with one 10
minute wash of the wells with 2X SSC/0.1% SDS (Sigma-Aldrich, USA), followed by 1 min wash with 0.2X SSC and finally 0.1X SSC was performed. Cleavage of probes from the surface was performed
in the next steps and not during in situ cDNA synthesis. The reaction mix consisted of 1.1X Second strand buffer (ThermoFisher Scientific, USA), 0.1 mM dNTPs and 1 U/µl USER enzyme (NEB,
USA). 75 µl of the mix was added and incubated for 3 h at 37 °C. The released material was collected in a new 96-well PCR plate (Eppendorf, Germany) by aspirating 70 µl of the released
material. SM-OMICS LIBRARY PREPARATION (2) Upon initiating the Agilent Bravo form the user was prompted to select either: 1, 2, 3, 4, 6 or 12 columns of the 96-well plate to run. Two
positions on the Bravo deck had Peltier thermal stations (4–95 °C) in the standard 96-well format. A reagent plate was prepared for robotic aspiration, transfer and dispensing of reagents.
First, single-stranded cDNA was made to double-stranded material using 5 µl of the reaction mix (2.7X First strand buffer, 3.7 U/µl DNA polymerase I and 0.2 U/µl Ribonuclease H (all from
ThermoFisher Scientific, USA)) for 2 h at 16°C. Thereafter, the material was blunted by the addition of 5 µl of 3U/µl T4 DNA polymerase (NEB, USA) for 20 min at 16 °C. The reaction was
stopped by addition of Invitrogen UltraPure 0.5 M EDTA (pH 8.0, ThermoFisher Scientific, USA) to a final concentration of 20 mM. The material was then purified using Ampure XP (Beckman
Coulter, USA) at a bead to cDNA ratio of 1:1. Next, 27.8 µl of the T7 reaction mix (46.2 mM rNTPs, 1.5X T7 reaction buffer, 1.54 U/µl SUPERaseIN inhibitor and 2.3 U/µl T7 enzyme; all from
ThermoFisher Scientific, USA) was added and sealed with 40 µl of Vapor-Lock oil (Qiagen, Germany) for an overnight 14 h incubation at 37 °C. After incubation, 2.1 µl of nuclease-free water
(ThermoFisher Scientific) was added and the Vapor-Lock was removed, followed by a bead cleanup with RNAclean Ampure XP beads (Beckman Coulter, USA) at a ratio of 1.8:1 of beads:aRNA. The
material was then assessed with a Bioanalyzer RNA 6000 Pico kit (Agilent Technologies, USA). 8 µl of the eluted 10 µl aRNA was transferred into a new 96-well PCR plate (Eppendorf, Germany).
SM-OMICS LIBRARY PREPARATION (3) 2.5 µl of either 3 µM (standard) or 15 µM aRNA adapters (efficient) [rApp]AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC[ddC] were added to 8 µl of aRNA. The reaction
was then incubated at 70 °C in a PCR machine for 2 min and immediately chilled on wet ice. The user then again selected the number of columns they wished to run. 4.5 µl T4 RNA ligation mix
(3.3X T4 RNA ligase buffer, 66U/µl truncated T4 ligase 2 and 13U/µl murine RNAse inhibitor (all from NEB, USA)) were added to the aRNA/adapter solution. The ligation reaction took place at
25 °C for 1 h (standard) or 3 h (efficient). For the SM-Omics protocol, the ligation reaction was performed for 3 h in the presence of 15 µM aRNA adapters. The ligation was followed by an
Ampure XP (Beckam Coulter, USA) bead purification at a ratio of 1.8:1 bead:cDNA. Elution volume was 12 µl. After bead purification, 2 µl of a primer and dNTP mix (1:1 v/v of either 20 µM or
40 µM GTGACTGGAGTTCAGACGTGTGCTCTTCCGA and 10 mM dNTPs) were added to the ligated samples. For the SM-Omics protocol, 40 µM primer amount was added using the same volumes. Then, the samples
were sealed with 40 µl Vapor-Lock (Qiagen, Germany) and heated to 65 °C for 5 min. The Vapor-Lock was thereafter removed and 8 µl of reverse transcription mix were added (2.5X First strand
buffer, 13 mM DTT, 5 U/µl RNaseOUT and 25 U/µl Superscript III; all from Thermo Fisher Scientific, USA), with the addition of 40 µl Vapor-Lock to reseal the reaction. The samples were
incubated at 50 °C for 1 h. 10 µl of nuclease-free water was added followed by a final Ampure XP bead purification at 1.7:1 bead:cDNA ratio with a final elution of 10 µl nuclease-free water.
STAINING TISSUES WITH OLIGONUCLEOTIDE-CONJUGATED ANTIBODIES As described above, the fresh frozen tissue was placed on the spatial array slide and fixed at RT, followed by antibody
incubations at 4 °C. First, tissues were blocked and permeabilized as described above. This was followed by a series of 3 washes in 1X PBS and a last wash that was incubated for 2 min. After
discarding the wash, oligonucleotide-conjugated antibodies and fluorescently labeled antibodies (Biolegend, USA) were both added at a 1:100 dilution in the same buffer as in the initial
permeabilization step and incubated for 1 h. The tissue was then washed and the antibody conjugates fixed to the array surface in 4% PFA (Sigma-Aldrich, USA). Tissues were then fluorescently
imaged and SM-Omics libraries created. The following steps were added in the library preparations to ensure collection of spatially DNA-barcoded antibody tags. First, cDNA synthesis was
performed in situ under the same conditions as described above. Next, second strand synthesis was also performed as described followed by an Ampure XP bead clean up as according to
manufacturer’s instructions. During this clean up, material that would otherwise have been discarded after binding to the beads in standard SM-Omics library preparations, was saved and
represented a population of spatially DNA-barcoded antibody tags. This elute contained short products that required a bead clean up procedure as well, where a 1.4X bead-to-material ratio was
used and the final product eluted in 50 µL EB (Qiagen, Germany). This material was then indexed for Illumina sequencing using Small RNA Illumina indexes in a KAPA indexing reaction as
described in _Quantification, indexing and sequencing_. MANUAL ST LIBRARY PREPARATION Manual library preparation was performed as described in Salmén et al.47 and included the same
experimental steps as the robotic library preparation protocol, but performed manually, incubations took place in a PCR System Eppendorf Mastercycler (Eppendorf, Germany) and instead of
Vapor-Lock, reactions were sealed using MicroAmp Optical 8-Cap Strips (ThermoFisher Scientific, USA). The manual procedure also included the following deviations from the robotic library
preparation: T7 reaction mix of 18.6 µl was used and 1.4 µl of nuclease-free water was added after the 14 h incubation. MANUAL VISIUM PREPARATION Cortical tissues from an adult mouse brain
were cryosectioned at 10 µm thickness and placed on Visium capture areas. The protocol was followed as in the Visium Spatial Gene Expression User Guide CG000239 Rev B as provided by 10X
Genomics. QUANTIFICATION, INDEXING AND SEQUENCING qPCR library quantification and indexing were performed as described in Salmén et al.47. The indexed SM-Omics cDNA libraries were diluted
with 40 µl of nuclease-free water to allow for a final library bead cleanup with 0.8:1 ratio Ampure XP beads to PCR products, according to the manufacturer’s protocol. Final elution was done
in 16 µl EB (Qiagen, Germany). Individual libraries’ fragment lengths and concentrations were evaluated on a Bioanalyzer HS (Agilent Technologies, USA) or DNA1000 Tapestation (Agilent
Technologies, USA) and DNA HS Qubit assays (ThermoFisher Scientific, USA), respectively. Samples were then diluted to the desired concentration for sequencing (~1.08 pM final for NextSeq
sequencing with 10% PhiX) and sequenced 27–30nt in the forward read and 55–58nt in the reverse read. For antibody tags, the final clean-up was performed at 0.9:1 ratio of beads to PCR
products and elution again done in 16 µl EB (Qiagen, Germany). Samples were diluted to 8pM final concentration before sequencing on an Illumina Miseq (2 × 25nt). STATISTICS AND
REPRODUCIBILITY RAW READS PROCESSING AND MAPPING ST, SM-Omics, Visium or antibody tag fastq reads were generated with bcl2fastq2. ST Pipeline59 v.1.7.6 was used to demultiplex the spatial
barcodes and collapse duplicate UMI sequences for ST, SM-Omics and Visium. In short, 5nt trimmed R2 was used for mapping to the mouse genome (GRCm38 primary assembly available at
https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/) using STAR (v2.6.0)60. After that, mapped reads were annotated using HTseq-count (v0.11.4)61 using the m11 gtf file
(https://www.gencodegenes.org/mouse/release_M11.html). To collapse UMIs, the annotated reads needed to first be connected to a spatial barcode using a TagGD59,62 (v0.3.6) demultiplexer
(k-mer 6, mismatches 2). Then, UMIs mapping to the same transcript and spatial barcode were collapsed using naive clustering with one mismatch allowed in the mapping process. The output file
was a genes-by-barcode matrix that was used in all further processing steps. To map antibody tags to their respective spatial barcodes, we used the tag quantification pipeline originally
developed for CITE-Seq (v.1.4.3) available at https://github.com/Hoohm/CITE-seq-Count. The pipeline was run with default parameters (maximum Hamming distance of 1). We additionally provided
the spatial barcodes and corrected the spatial mapping (1 mismatch) for a total of 1007 different barcodes. SPOTTER: AUTOMATED IMAGE REGISTRATION FOR SPATIAL TRANSCRIPTOMICS ARRAYS For
efficient processing, HE images were scaled to approximately 500 × 500 pixels using the imagemagick (https://imagemagick.org/index.php) mogrify command. Other image operations as mentioned
below were performed using the R package imager (http://dahtah.github.io/imager/) unless specified differently. In order to reconstruct the positions of all ST spots, visible (_i.e_., not
covered by the tissue section) barcode (x,y) spots were registered through blob detection and then refined by keeping only those blobs (i.e. potential grid points) that were likely to be
part of a regular grid. Blob detection refers to finding circular features of a predefined size in the image. To prepare the H&E image for blob detection, the tissue section was masked
generously through 10% quantile thresholding in a user-defined color channel as obtained through the function imsplit. The borders of the resulting image were cropped four pixels from each
of the four image borders in order to remove any abnormality or border effects that might interfere with blob detection. For blob detection, we first blurred the cropped image isotropically
using the function isoblur with sigma = 3 and then computed the image hessian (function imhessian). This allowed us to detect probable blob centers using the function dilate_square with size
= 3 and the function pad with nPix = 4 and pos = −1. Blob centers (i.e., potential grid points) that were likely part of a regular grid structure were selected by calculating the x and y
distances between all detected blob centers. Those blob centers that based on their 8 nearest neighbors had a high (empirically determined) combined grid score (metric based on distance and
grid angle between the neighboring centers) were kept. A regular grid was then fitted to these potential grid points using a custom optimizer built around the function nlminb of the R
package stats, that minimizes the distance of potential grid points to the suggested regular grid, while assuming 90° angles and 42 grid points per row and column. This first, rough grid
initialized an iterative process in which the 0.1% potential grid points that least fit the grid were removed in each iteration, the number of grid points per row and column were updated
accordingly, and a new grid was fitted until the target number of grid points per row (here 35) and column (here 33) was reached. The grid design and target number of rows and columns is
fully adjustable in SpoTteR. Finally, those grid points that overlapped the tissue sections were identified by building a mask that represented the tissue area and registering all grid
points that were present in this mask. To build this mask, we calculated the mean and standard deviation of the background intensity based on the first 20 pixels from the image border,
because no tissues were expected in that area. Pixels with an intensity greater than the mean background intensity adjusted for its standard deviation were set to 1 (likely tissue) and those
below or equal the mean background intensity adjusted for its standard deviation were set to 0 (likely background), creating the primary tissue mask. Complementarily, a background mask was
created by selecting all likely background pixels using the function px.flood with sigma = 0.1 and removing those pixels from the primary tissue mask in order to remove dark artefacts. Two
final rounds of isotropic blurring using the function isoblur with sigma = 10 and sigma = 1 in combination with intensity thresholding enhanced the detection of weakly colored tissue regions
such areas around the tissue edges. In order to further accommodate atypical tissue coloring, bubbles, and smears present as imaging artifacts, we introduced a parameter to specify the
usage of the green color channel instead of the red color channel for tissue detection, which exploits the observation that smears and H&E staining artefacts often lead to spurious pink
coloring, which is especially strong in the red channel. To address bubbles, another common image artefact, we introduced a parameter that allows the creation of an additional bubble mask
based on all three color channels that specifically identifies bubbles as features that have roughly the same low intensity in all three color channels as they are typically dark gray or
even black in color. The thus identified likely bubble pixels are then also removed from the tissue mask. These two additional (TRUE/FALSE) parameters enable to easily process data from
tissues of various degrees of coloration and bubble artefacts. Finally, an intermediate report notifies the user of irregularities in the automatic alignment process and allows for visual
inspection. The output.tsv file contained barcode spots (x,y) as centroid pixel coordinates of the detected grid, as well as a TRUE/FALSE value, set as TRUE if the barcode spot was detected
as under the tissue section area. SPOTTER INTEGRATION WITH ST PIPELINE AND QUALITY CONTROL REPORTING The following steps integrate the output from the automated image alignment steps with
the output gene-by-barcode expression file as produced by the ST Pipeline v.1.7.6. The barcode (x,y) spots approximated as under the tissue section were used for subsetting the ST Pipeline
gene-by-barcode file. Then, the original H&E images were downscaled and cropped using the following imagemagick commands: convert HE_image.jpg -crop width“x“height+xa+ya; where width and
height represented the Euclidean lengths between (x,y) grid detected barcode spots (33,35), (1,35) and (1,35), respectively. xa and ya were described as the centroid pixel coordinates of
the grid point (33,35). The cropped H&E image was then rotated as follows: mogrify -flop -flip HE_image.jpg and this image was then used as input to the QC reporting system and for the
GUI annotation tool. A final quality control (QC) report was created when running SpoTteR. All code for running image registration and QC reporting with SpoTteR has been made available at:
https://github.com/klarman-cell-observatory/SpoTteR. COMPARISON OF SPOTTER _VS_. ST SPOT DETECTOR VS. MANUAL ALIGNMENT To be able to compare the automated image processing developed here to
that of manually processed images, we acquired an additional image of the ST array area after the experiment was performed and the tissue had been removed from the array surface. Briefly,
complementary and Cy3 labeled oligonucleotides (IDT, USA) were diluted in 2X SSC with 0.05% SDS to a final concentration of 1 µM. 50 µl of the diluted solution was added to the array surface
and incubated with shaking (50 rpm) for 10 min at RT. This was followed by washing the slide in 4XSCC with 0.1% SDS and 0.2X SSC. The array frame and all ST barcode positions had then
efficiently been labeled and acquired on the same imaging system as described. All input images in the following comparisons were the same approximate input sizes and resolution. The ST spot
detector tool previously developed48 uses the H&E and Cy3 images as input. Due to its intrinsic scaling factor and input image size requirements, initial pre-processing of both images
was needed, such that images be linearly downscaled to 30% of their original size and both images individually cropped to represent the same FOVs as collected during the imaging step.
However, cropping was only needed if the user did not have the possibility to automatically acquire the same FOVs using the same starting (x,y) positions. For manual alignment, we used Adobe
Photoshop for initial pre-processing, same as in the previous step. Both the H&E and Cy3 acquired images were downscaled to 30% of their original size, rotated 180 degrees and aligned
to the same starting (x,y) pixel coordinates. This was followed by cropping both images along the middle of the first and last row and column. The tissue boundaries were detected using the
magic wand function (32px) and the selection subtracted in the Cy3 image. Spots boundaries were again detected using the same magick wand function and the background noise cleaned up using
the bucket fill function (250px) in a grayscale image. This grayscale image was further used in Fiji63 to detect the centroid coordinates of each ST barcode spot. Following Fiji processing,
we translated (x,y) pixel centroid coordinates to ST barcode spot coordinates (as given during the demultiplexing step in the ST pipeline). For SpoTteR input, we only provided the original
H&E imaged as acquired by the imaging system with no GUI-based preprocessing. For speed comparisons, total time needed for preprocessing steps was measured first. For manual processing,
the pre-processing steps included alignment of the H&E and Cy3 images with Adobe Photoshop 2019 and creation of an ST array spots files. For ST Detector pre-processing time, we only took
into consideration the time needed to open the same images in Adobe Photoshop, downscale them to 30% size and crop them the same size without any other image handling processes performed.
For SpoTteR, preprocessing included the downscaling step performed with imagemagick and incorporated into the workflow. Processing steps were then performed and time was measured as
described before. Total speed was considered as 1/t [s−1] where t represents the sum of time needed for both the pre-processing and processing steps. False positive and negative rates were
calculated as percentage of spots present or absent in SpoTteR or ST Detector as compared to manually processed spot coordinates. ESTIMATING LATERAL DIFFUSION Two consecutive mouse cortex
fresh frozen sections were processed. One was processed manually as described earlier47 while the other was processed using our devised robotic liquid handling setup. For these tests, we
created poly(d)T arrays in-house according to manufacturer’s instructions (Codelink, Surmodics, USA) using amine-activated slides. The surface area covered with poly(d)T probes was 6x6mm.
Both the H&E and gene activity Cy3 images were processed in Fiji63. First, in order to detect the nuclear boundaries of cells chosen at random throughout the tissue, we drew a line
(Straight > Freehand line) through each visible nucleus (_n_ = 50). Secondly, we collected pixel intensities and distances reaching through each of the chosen nuclei and its surrounding
area (Analyze > Plot Profile). To distinguish nuclear boundaries in the collected intensity _vs_. distance data, we first fit a 5th degree polynomial of the curve. Then, we found local
minima and maxima in each curve and determined cell boundaries as local minima present at above 10% signal intensity of the local maximum value for each curve. After cell boundaries were
defined, we repeated the process using the Cy3 fluorescent gene activity image. Finally, we measured the distance between the detected Cy3 and nuclear signals for each selected cell. Left
and right cell boundaries representing opposite sides of each cell were used in the estimate in each condition. A 0.1728 pixel to distance conversion ratio was used to transform pixels to
micrometers reported in this paper. If a diffusion distance measure was scored as negative it implied that the Cy3 signal was contained within the detected cell boundaries, and positive if
outside those same boundaries. ESTIMATING REPRODUCIBILITY OF SM-OMICS IN SITU REACTIONS Scikit-image64 was used to process the H&E and respective fluorescent gene expression images.
First, a grayscale fluorescent image was smoothed using a Gaussian filter (sigma = 0.01). Then, we applied morphological reconstruction by dilating the image edges through filtering its
regional maxima. This enabled us to create a background image value that could be subtracted from the original image and used in further analysis. Then, we created an elevation map with a
Sobel filter to mask the elevated points. This image could then be used in a tissue (i.e., object) detection step using watershedding. The inverted tissue boundaries were subtracted from the
detected fluorescent tissue gene expression signals and used in all further analysis. The means of the fluorescent signals were compared using a two-sided Wilcoxon’s rank-sum test. If the
expected signal-to-noise ratio between the detected gene expression signature and background signals was less than 3:1 new tissue optimizations are recommended. ANNOTATION PATTERNS THROUGH
MANUAL IMAGE ANNOTATION AND REGISTRATION To manually annotate tissue images based on their H&E features, we used a previously adapted graphical and cloud-based user interface26. We
assigned each ST (x,y) coordinate with one or more regional tags. The region names used to annotate MOB were: granular cell layer (GR), outer plexiform layer (OPL), mitral layer (MI),
internal plexiform layer (IPL) and glomerular layer (GL) and to annotate mouse cortex were: cerebral nuclei (CNU), cortical subplate (CTXsp), fiber tracts, hippocampal formation (HIP),
hypothalamus (HY), isocortex (ISOCTX), midbrain (MB), piriform area (PIR) and thalamus (TH). For annotating spleen, we used four major areas: red pulp, B-follicle, marginal zone and
periarteriolar lymphoid sheaths (PALS). To overlay tissue images through an image registration task, we used centroids of each annotated region as anchor points in the image translation and
rotation tasks, as previously described32. This allowed us to display the data in a common coordinate system and to highlight genes and annotation areas of interest. COMPARISONS BETWEEN
SPATIAL GENE EXPRESSION PROFILES For comparisons between the SM-Omics and ST datasets, reads were first downsampled to the same saturation level before invoking the ST pipeline mapper,
annotator and counter run to receive UMIs per spatial (x,y) barcode. Depending on sequencing depth, a gene was counted as expressed if the corresponding transcript was present in more than
10−6 of the sequencing depth. The total count over all spots per gene and sample were then normalized65. Spearman’s correlation coefficient between the average and normalized samples was
calculated using Scipy v1.2.066. To compare the performance of Visium and SM-Omics, we sequenced both libraries to an average depth of ~65 million paired end reads. For Visium, we sequenced
29 nt in the forward and 43 nt in the reverse read. Reads were downsampled to the same saturation level. Both datasets were processed using the ST pipeline as described above. Conventional
GTF files used in the annotation step with HTseq-count were converted so that all transcript features now carried an exon tag used in counting transcripts. UMI collapsing was done using a
naive approach and allowing for 1 low quality base present in either of the datasets. Unique molecular identifiers per measurement were calculated as described earlier. To visualize the
counts data per condition, total numbers of detected genes or UMIs were plotted as violin plots and summarized mean values for all replicate libraries overlayed as dotplots; similar as
presented in Lord et al.67. To compare between different spatial RNA-seq protocol versions, we followed an approach similar to that previously described in Svensson et al.68. Raw data were
first processed as described in the _Saturation curve generation_ section, and each replicate (at least _n_ = 3) from each condition (i.e., spatial RNA-seq protocol version) was represented
by the counts mean67 at each of 9 different saturation points. Following processing, summarized counts data in each comparison were first scaled [0,1] and then used to estimate a generalized
linear mixed model (glmm). We used a glmm (R package glmmTMB v1.1.1) modeled as a proportional binomial logit response between counts, protocol version (fixed effect) and replicate (random
effect). Log proportions of annotated reads were used as offsets in the model. All glmm estimates were performed using the R stats package (v4.0.1) and Wald’s p-values reported. SATURATION
CURVE GENERATION Number of unique molecules was calculated by subsampling the same proportion of mapped and annotated reads from each sample. First, each library was randomly down-sampled to
three sequencing saturation points (defined as percentage of raw reads in a library) and numbers of UMIs or unique genes and annotated reads in a sample collected after running the ST
Pipeline v.1.7.6 as described in the _Raw reads processing and mapping_ section above. Using this information, we could solve the Lineweaver–Burk equation and accurately estimate the number
of raw reads _R_ in each sample _s_ that are needed to reach a certain saturation level _S_ in a given library: $${R}_{s}=\frac{{S}_{s}\times {K}_{M}}{{V}_{{\max }}-{S}_{s}},$$ (1) where
_V_max is the maximum saturation point and _K__M_ represents the number of raw reads at half of _V_max After randomly down-sampling all the libraries to the same library saturation, we
considered this our maximum saturation point (100%) in all comparisons and sampled a total of 9 different points (0.001–100%) to be included in the saturation curve plots presented in this
paper. QUANTITATIVE IMMUNOFLUORESCENCE PROFILES PER SM-OMICS SPOT First, we trained a random forest classifier using the Ilastik69 (v1.3.3) framework to extract probabilities of the positive
class assignment ie. positive antibody signals from our IF mouse brain images. Separate classifiers were trained to each antibody used and a total of ~10 images with at least 10 fields of
view were used in the training process. In each classifier, we used two labels for classification: signal and background. Respective full-sized fluorescent microscopy images were then
processed and output probabilities used in the following steps. For spleen data, raw fluorescent images were used as input in the following steps. First, images were processed as described
in _Estimating reproducibility of SM-Omics_ in situ _reactions_. Calculated background was removed from each image, signal boundaries estimated using watershedding followed by creating a
binary mask image. This mask was then overlaid with the original fluorescent image and this image was then used in all following steps. To quantify the fluorescent signal intensities per ST
spot, the image was cropped into a 33 × 35 matrix creating smaller patches; each patch sized at ±1% image from the centroid of each ST spot. Finally, the intensity from each spot area was
calculated as the sum of the fluorescent signal detected in that spot patch. SPATIAL GENE AND ANTIBODY-BASED EXPRESSION ANALYSIS Statistical analysis of the spatial gene and antibody tag
expression data was performed using Splotch’ one- or two-level hierarchical model as previously described32. In short, the model captures spatial expression in anatomical regions while
accounting for experimental parameters such as, in our case, different animals, and calculates gene or antibody expression estimates for each single gene or antibody in each annotated
spatial spot. To find targets which were differentially expressed in an annotated morphological region, we computed a one-_vs_-all comparison and took those values with a positive log
Bayesian factor (BF). Posterior probabilities presented hereinafter normalized expression estimates and were used throughout the analyses presented. For scaling per annotated region,
normalized expression values were first grouped by annotated region and then scaled from 0 to 1 within each sample. The correlation between gene expression and fluorescent signal was
calculated in the same way, but the fluorescent signal matrix, prepared as explained in _Calculating quantitative immunofluorescence profiles per SM-Omics spot_, was used instead of the
antibody tag counts matrix. COMPARISON TO ALLEN BRAIN ATLAS DATA To validate our findings, we downloaded in situ hybridization (ISH) gene expression data from the Allen Brain Atlas50
(https://mouse.brain-map.org/) with Image Credit: Allen Institute for Brain Science. The following gene expression images were used from the ABA as denoted with appropriate image and
experiment identifiers: _CTGF_ 478 [https://mouse.brain-map.org/experiment/show/79556634], _CAMK4_ 474 [https://mouse.brain-map.org/experiment/show/75038464], _LANCL3_ 474
[https://mouse.brain-map.org/experiment/show/73925716], _CBLN4_ 476 [https://mouse.brain-map.org/experiment/show/72283804], _NR2F2_ 466 and 250
[https://mouse.brain-map.org/experiment/show/112646890], _NRSN1_ 478 [https://mouse.brain-map.org/experiment/show/71358557], _NOS1AP_ 472
[https://mouse.brain-map.org/experiment/show/77280574], _CDH23_ 469 [https://mouse.brain-map.org/experiment/show/72283805], _PRSS12_ 474
[https://mouse.brain-map.org/experiment/show/71836879], _CABP7_ 253 [https://mouse.brain-map.org/experiment/show/73930835], _SEMA4G_ 266
[https://mouse.brain-map.org/experiment/show/71587856], _DKKL1_ 237 [https://mouse.brain-map.org/experiment/show/70634395], _SLC17A6_ 272
[https://mouse.brain-map.org/experiment/show/73818754] and _PENK_ 262 [https://mouse.brain-map.org/experiment/show/74881286]. For comparisons in MOB samples, we used the following regions
from ABA: GL, GR, MI and OPL. For comparison in cortex samples, we used the following regions from ABA: piriform-amygdalar area (PAA), postpiriform transition area (TR) in addition to CNU,
CTXsp, HIP, HY, ISOCTX, MB and TH. Prior to enrichment analysis, genes found in PAA, TR and PIR in ABA were merged into one region name: PIR. We filtered genes with fold change >1 and
expression threshold >2.5 in ABA and compared to genes with positive fold change and log(BF) in our Splotch data and computed a one-sided Fisher’s exact test using Scipy v1.2.066. FDR was
estimated using the Benjamini-Hochberg70 procedure. Heatmaps denoting regions present in both conditions were plotted. One of the top most differentially expressed genes in both SM-Omics
and ABA was chosen from each region and its expression visualized. A reference ST dataset24 was also analyzed using Splotch with the same settings as used for SM-Omics, visualized and
compared to SM-Omics. To create correlations between ABA expression patterns and SM-Omics, Visium and ST expression patterns, normalized expression data was first grouped by annotated region
and then scaled from 0 to 1 within each sample. To compare SM-Omics and ST, we compared top genes per MOB region: _NRSN1_, _NOS1AP_, _CDH23_ and _PRSS12_. To compare SM-Omics and Visium, we
compared top genes per mouse brain cortex as found in ABA: _ADORA2A_, _CABP7_, _SLC6A11_, _IER5_, _SLC17A6_ and _GREM2_. REPORTING SUMMARY Further information on research design is
available in the Nature Research Reporting Summary linked to this article. DATA AVAILABILITY Raw sequencing data is available at NCBI’s Bioproject under accession PRJNA797464. All processed
and source data generated in this study have been deposited in the Single Cell Portal under accession code SCP979. All other relevant data supporting the key findings of this study are
available within the article and its Supplementary Information files. CODE AVAILABILITY All code is on GitHub at https://github.com/klarman-cell-observatory/sm-omics. REFERENCES * Jovanovic,
M. et al. Immunogenetics. Dynamic profiling of the protein life cycle in response to pathogens. _Science_ 347, 1259038 (2015). Article PubMed PubMed Central CAS Google Scholar *
Rabani, M. et al. High-resolution sequencing and modeling identifies distinct dynamic RNA regulatory strategies. _Cell_ 159, 1698–1710 (2014). Article CAS PubMed PubMed Central Google
Scholar * Chick, J. M. et al. Defining the consequences of genetic variation on a proteome-wide scale. _Nature_ 534, 500–505 (2016). Article ADS CAS PubMed PubMed Central Google
Scholar * Zhang, B. et al. Proteogenomic characterization of human colon and rectal cancer. _Nature_ 513, 382–387 (2014). Article CAS PubMed PubMed Central Google Scholar * Stoeckius,
M. et al. Simultaneous epitope and transcriptome measurement in single cells. _Nat. Methods_ 14, 865–868 (2017). Article CAS PubMed PubMed Central Google Scholar * Zheng, G. X. Y. et
al. Massively parallel digital transcriptional profiling of single cells. _Nat. Commun._ 8, 14049 (2017). Article ADS CAS PubMed PubMed Central Google Scholar * Macosko, E. Z. et al.
Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. _Cell_ 161, 1202–1214 (2015). Article CAS PubMed PubMed Central Google Scholar * Klein, A.
M. et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. _Cell_ 161, 1187–1201 (2015). Article CAS PubMed PubMed Central Google Scholar * Habib, N.
et al. Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. _Science_ 353, 925–928 (2016). Article ADS CAS PubMed PubMed Central Google Scholar * Habib, N.
et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. _Nat. Methods_ 14, 955–958 (2017). Article CAS PubMed PubMed Central Google Scholar * Slyper, M. et al. A single-cell
and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. _Nat. Med._ 26, 792–802 (2020). Article CAS PubMed PubMed Central Google Scholar * Darmanis, S. et al. Simultaneous
multiplexed measurement of RNA and proteins in single cells. _Cell Rep._ 14, 380–389 (2016). Article CAS PubMed Google Scholar * Hanahan, D. & Coussens, L. M. Accessories to the
crime: functions of cells recruited to the tumor microenvironment. _Cancer Cell_ 21, 309–322 (2012). Article CAS PubMed Google Scholar * Bodenmiller, B. Multiplexed epitope-based tissue
imaging for discovery and healthcare applications. _Cell Syst._ 2, 225–238 (2016). Article CAS PubMed Google Scholar * Chen, K. H., Boettiger, A. N., Moffitt, J. R., Wang, S. &
Zhuang, X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. _Science_ 348, aaa6090 (2015). Article PubMed PubMed Central CAS Google Scholar * Lubeck,
E., Coskun, A. F., Zhiyentayev, T., Ahmad, M. & Cai, L. Single-cell in situ RNA profiling by sequential hybridization. _Nat. Methods_ 11, 360–361 (2014). Article CAS PubMed PubMed
Central Google Scholar * Eng, C.-H. L. et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. _Nature_ 568, 235–239 (2019). Article ADS CAS PubMed PubMed Central
Google Scholar * Lee, J. H. et al. Highly multiplexed subcellular RNA sequencing in situ. _Science_ 343, 1360–1363 (2014). Article ADS CAS PubMed PubMed Central Google Scholar *
Goltsev, Y. et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. _Cell_ 174, 968–981.e15 (2018). Article CAS PubMed PubMed Central Google Scholar * Keren,
L. et al. A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. _Cell_ 174, 1373–1387.e19 (2018). Article CAS PubMed
PubMed Central Google Scholar * Codeluppi, S. et al. Spatial organization of the somatosensory cortex revealed by osmFISH. _Nat. Methods_ 15, 932–935 (2018). Article CAS PubMed Google
Scholar * Moffitt, J. R. et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. _Science_. 362, 749–750 (2018). Article CAS Google Scholar *
Ke, R. et al. In situ sequencing for RNA analysis in preserved tissue and cells. _Nat. Methods_ 10, 857–860 (2013). Article CAS PubMed Google Scholar * Ståhl, P. L. et al. Visualization
and analysis of gene expression in tissue sections by spatial transcriptomics. _Science_ 353, 78–82 (2016). Article ADS PubMed CAS Google Scholar * Rodriques, S. G. et al. Slide-seq: a
scalable technology for measuring genome-wide expression at high spatial resolution. _Science_ 363, 1463–1467 (2019). Article ADS CAS PubMed PubMed Central Google Scholar * Vickovic,
S. et al. High-definition spatial transcriptomics for in situ tissue profiling. _Nat. Methods_ 16, 987–990 (2019). Article CAS PubMed PubMed Central Google Scholar * Wang, X. et al.
Three-dimensional intact-tissue sequencing of single-cell transcriptional states. _Science_. 361, 328–329 (2018). Article Google Scholar * Maynard, K. R. et al. Transcriptome-scale spatial
gene expression in the human dorsolateral prefrontal cortex. _Nat. Neurosci._ 24, 425–436 (2021). Article CAS PubMed PubMed Central Google Scholar * Thrane, K., Eriksson, H., Maaskola,
J., Hansson, J. & Lundeberg, J. Spatially resolved transcriptomics enables dissection of genetic heterogeneity in stage III cutaneous malignant melanoma. _Cancer Res_ 78, 5970–5979
(2018). CAS PubMed Google Scholar * Asp, M. et al. Spatial detection of fetal marker genes expressed at low level in adult human heart tissue. _Sci. Rep._ 7, 12941 (2017). Article ADS
PubMed PubMed Central CAS Google Scholar * Asp, M. et al. A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. _Cell_ 179, 1647–1660.e19 (2019).
Article CAS PubMed Google Scholar * Maniatis, S. et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. _Science_ 364, 89–93 (2019). Article ADS CAS
PubMed Google Scholar * Berglund, E. et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. _Nat. Commun._ 9, 2419 (2018). Article ADS
PubMed PubMed Central CAS Google Scholar * Berglund, E. et al. Automation of spatial transcriptomics library preparation to enable rapid and robust insights into spatial organization of
tissues. _BMC Genomics_ 21, 298 (2020). Article CAS PubMed PubMed Central Google Scholar * Jemt, A. et al. An automated approach to prepare tissue-derived spatially barcoded
RNA-sequencing libraries. _Sci. Rep._ 6, 37137 (2016). Article ADS CAS PubMed PubMed Central Google Scholar * Vickovic, S. et al. Massive and parallel expression profiling using
microarrayed single-cell sequencing. _Nat. Commun._ 7, 13182 (2016). Article ADS CAS PubMed PubMed Central Google Scholar * Lein, E., Borm, L. E. & Linnarsson, S. The promise of
spatial transcriptomics for neuroscience in the era of molecular cell typing. _Science_ 358, 64–69 (2017). Article ADS CAS PubMed Google Scholar * Gerdes, M. J. et al. Highly
multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. _Proc. Natl Acad. Sci. USA_ 110, 11982–11987 (2013). Article ADS CAS PubMed PubMed Central Google
Scholar * Lin, J.-R., Fallahi-Sichani, M. & Sorger, P. K. Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. _Nat. Commun._ 6, 8390
(2015). Article ADS CAS PubMed Google Scholar * Angelo, M. et al. Multiplexed ion beam imaging of human breast tumors. _Nat. Med._ 20, 436–442 (2014). Article CAS PubMed PubMed
Central Google Scholar * Giesen, C. et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. _Nat. Methods_ 11, 417–422 (2014). Article CAS
PubMed Google Scholar * Merritt, C. R. et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. _Nat. Biotechnol._ 38, 586–599 (2020). Article CAS PubMed Google
Scholar * Schulz, D. et al. Simultaneous multiplexed imaging of mRNA and proteins with subcellular resolution in breast cancer tissue samples by mass cytometry. _Cell Syst._ 6, 531 (2018).
Article CAS PubMed PubMed Central Google Scholar * 10x Genomics. https://www.10xgenomics.com/products/spatial-proteomics. * Fisher, S. et al. A scalable, fully automated process for
construction of sequence-ready human exome targeted capture libraries. _Genome Biol._ 12, R1 (2011). Article PubMed PubMed Central Google Scholar * Rohland, N. & Reich, D.
Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. _Genome Res_ 22, 939–946 (2012). Article CAS PubMed PubMed Central Google Scholar * Salmén, F.
et al. Barcoded solid-phase RNA capture for spatial transcriptomics profiling in mammalian tissue sections. _Nat. Protoc._ 13, 2501–2534 (2018). Article PubMed CAS Google Scholar * Wong,
K., Navarro, J. F., Bergenstråhle, L., Ståhl, P. L. & Lundeberg, J. ST Spot Detector: a web-based application for automatic spot and tissue detection for spatial Transcriptomics image
datasets. _Bioinformatics_ 34, 1966–1968 (2018). Article CAS PubMed Google Scholar * Äijö, T. et al. Splotch: Robust estimation of aligned spatial temporal gene expression data.
https://doi.org/10.1101/757096. * Lein, E. S. et al. Genome-wide atlas of gene expression in the adult mouse brain. _Nature_ 445, 168–176 (2007). Article ADS CAS PubMed Google Scholar *
Kang, H. et al. An important role of neural activity-dependent CaMKIV signaling in the consolidation of long-term memory. _Cell_ 106, 771–783 (2001). Article CAS PubMed Google Scholar *
Mann, A. P. et al. Identification of a peptide recognizing cerebrovascular changes in mouse models of Alzheimer’s disease. _Nat. Commun_. 8, 1403 (2017). Article ADS PubMed PubMed
Central CAS Google Scholar * TotalSeqTM. https://www.biolegend.com/en-us/totalseq. * Nagayama, S., Homma, R. & Imamura, F. Neuronal organization of olfactory bulb circuits. _Front.
Neural Circuits_ 8, 98 (2014). Article PubMed PubMed Central Google Scholar * Zeisel, A. et al. Molecular architecture of the mouse nervous system. _Cell_ 174, 999–1014.e22 (2018).
Article CAS PubMed PubMed Central Google Scholar * Stickels, R. R. et al. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. _Nat. Biotechnol._ 39,
313–319 (2021). Article CAS PubMed Google Scholar * Chen, A. et al. Large field of view-spatially resolved transcriptomics at nanoscale resolution. Preprint at
https://doi.org/10.1101/2021.01.17.427004 (2021). * Su, G. et al. Spatial multi-omics sequencing for fixed tissue via DBiT-seq. _STAR Protoc._ 2, 100532 (2021). Article PubMed PubMed
Central Google Scholar * Navarro, J. F., Sjöstrand, J., Salmén, F., Lundeberg, J. & Ståhl, P. L. ST Pipeline: an automated pipeline for spatial mapping of unique transcripts.
_Bioinformatics_ 33, 2591–2593 (2017). Article PubMed CAS Google Scholar * Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. _Bioinformatics_ 29, 15–21 (2013). Article CAS
PubMed Google Scholar * Anders, S., Pyl, P. T. & Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. _Bioinformatics_ 31, 166–169 (2015). Article CAS
PubMed Google Scholar * Costea, P. I., Lundeberg, J. & Akan, P. TagGD: fast and accurate software for DNA Tag generation and demultiplexing. _PLoS ONE_ 8, e57521 (2013). Article ADS
CAS PubMed PubMed Central Google Scholar * Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. _Nat. Methods_ 9, 676–682 (2012). Article CAS PubMed
Google Scholar * van der Walt, S. et al. scikit-image: image processing in Python. _PeerJ_ 2, e453 (2014). Article PubMed PubMed Central Google Scholar * Svensson, V., Teichmann, S. A.
& Stegle, O. SpatialDE: identification of spatially variable genes. _Nat. Methods_ 15, 343–346 (2018). Article CAS PubMed PubMed Central Google Scholar * Jones, E., Peterson, P.
& Oliphant, T. SciPy: Open Source Scientific Tools for Python. _Scipy_ http://www.scipy.org/ (2001). * Lord, S. J., Velle, K. B., Mullins, R. D. & Fritz-Laylin, L. K. SuperPlots:
communicating reproducibility and variability in cell biology. _J. Cell Biol_. 219, e202001064 (2020). Article CAS PubMed PubMed Central Google Scholar * Svensson, V. et al. Power
analysis of single-cell RNA-sequencing experiments. _Nat. Methods_ 14, 381–387 (2017). Article CAS PubMed PubMed Central Google Scholar * Berg, S. et al. ilastik: interactive machine
learning for (bio)image analysis. _Nat. Methods_ 16, 1226–1232 (2019). Article CAS PubMed Google Scholar * Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a
practical and powerful approach to multiple testing. _J. R. Stat. Soc. Ser. B_ 57, 289–300 (1995). MathSciNet MATH Google Scholar Download references ACKNOWLEDGEMENTS We thank Ania
Hupalowska for making all graphical illustrations. We thank Tarmö Äijö for help with Splotch, Valentine Svensson for help with analysis and Theresa Ten Eyck for help with Bravo protocol
implementation. Work was supported by the Knut and Alice Wallenberg Foundation, the Royal Swedish Academy of Sciences and Swedish Society for Medical Research (S.V.), the Hans Werthén
Foundation (B.L), HFSP long term fellowship (LT000452/2019-L) (J.K.), DFG research fellowship (MA 9108/1-1) (S.M.), the Klarman Cell Observatory, the Manton Foundation, and HHMI (A.R.). S.V
was supported as a Wallenberg Fellow at the Broad Institute of MIT and Harvard. AUTHOR INFORMATION Author notes * O. Rozenblatt-Rosen & A. Regev Present address: Genentech, 1 DNA Way,
South San Francisco, CA, USA * These authors contributed equally: S. Vickovic, B. Lötstedt. AUTHORS AND AFFILIATIONS * Klarman Cell Observatory Broad Institute of MIT and Harvard, Cambridge,
MA, USA S. Vickovic, B. Lötstedt, J. Klughammer, S. Mages, Å Segerstolpe, O. Rozenblatt-Rosen & A. Regev * Department of Biology, Massachusetts Institute of Technology, Cambridge, MA,
USA S. Vickovic * New York Genome Center, New York, NY, USA S. Vickovic * Science for Life Laboratory, Department of Biochemistry and Biophysics, Stockholm University, Solna, Sweden S.
Vickovic * Science for Life Laboratory, Department of Gene Technology, KTH Royal Institute of Technology, Stockholm, Sweden B. Lötstedt * Department of Biological Engineering, Massachusetts
Institute of Technology, Cambridge, MA, USA B. Lötstedt * Howard Hughes Medical Institute and Koch Institute for Integrative Cancer Research, Department of Biology, Massachusetts Institute
of Technology, Cambridge, MA, USA A. Regev Authors * S. Vickovic View author publications You can also search for this author inPubMed Google Scholar * B. Lötstedt View author publications
You can also search for this author inPubMed Google Scholar * J. Klughammer View author publications You can also search for this author inPubMed Google Scholar * S. Mages View author
publications You can also search for this author inPubMed Google Scholar * Å Segerstolpe View author publications You can also search for this author inPubMed Google Scholar * O.
Rozenblatt-Rosen View author publications You can also search for this author inPubMed Google Scholar * A. Regev View author publications You can also search for this author inPubMed Google
Scholar CONTRIBUTIONS S.V. and A.R. designed the study and experiments; S.V. and B.L. performed the experiments with help from Å.S. for the automation testing steps; S.V., J.K. and S.M.
designed and implemented the automatic alignment and reporting tool; B.L. analyzed data with supervision from S.V.; O.R.R. helped plan spatial multi-omics experiments; S.V., B.L. and A.R.
wrote the manuscript with input from all the authors. All authors discussed the results. CORRESPONDING AUTHORS Correspondence to S. Vickovic or A. Regev. ETHICS DECLARATIONS COMPETING
INTERESTS A.R. is a founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas Therapeutics and until August 31, 2020 was a SAB member of Syros Pharmaceuticals, Neogene
Therapeutics, Asimov and ThermoFisher Scientific. From August 1, 2020, A.R. is an employee of Genentech. O.R.R. is a co-inventor on patent applications filed by the Broad Institute for
inventions related to single cell genomics. She has given numerous lectures on the subject of single cell genomics to a wide variety of audiences and in some cases, has received remuneration
to cover time and costs. O.R.R. is an employee of Genentech since October 19, 2020. S.V is an author on patents applied for by Spatial Transcriptomics AB (10X Genomics Inc). S.V. and A.R.
are co-inventors on PCT/US2020/015481 relating to this work. The remaining authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Quan
Nguyen, Tom Smith and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with
regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION REPORTING SUMMARY DESCRIPTION OF ADDITIONAL
SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 SUPPLEMENTARYDATA 2 SUPPLEMENTARY DATA 3 RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0
International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the
source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Vickovic, S., Lötstedt, B., Klughammer, J. _et al._ SM-Omics is an automated
platform for high-throughput spatial multi-omics. _Nat Commun_ 13, 795 (2022). https://doi.org/10.1038/s41467-022-28445-y Download citation * Received: 21 October 2020 * Accepted: 24 January
2022 * Published: 10 February 2022 * DOI: https://doi.org/10.1038/s41467-022-28445-y SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get
shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative