Association between SNPs and gene expression in multiple regions of the human brain
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

Identifying the genetic cis associations between DNA variants (single-nucleotide polymorphisms (SNPs)) and gene expression in brain tissue may be a promising approach to find functionally
relevant pathways that contribute to the etiology of psychiatric disorders. In this study, we examined the association between genetic variations and gene expression in prefrontal cortex,
hippocampus, temporal cortex, thalamus and cerebellum in subjects with psychiatric disorders and in normal controls. We identified cis associations between 648 transcripts and 6725 SNPs in
the various brain regions. Several SNPs showed brain regional-specific associations. The expression level of only one gene, PDE4DIP, was associated with a SNP, rs12124527, in all the brain
regions tested here. From our data, we generated a list of brain cis expression quantitative trait loci (eQTL) genes that we compared with a list of schizophrenia candidate genes downloaded
from the Schizophrenia Forum (SZgene) database (http://www.szgene.org/). Of the SZgene candidate genes, we found that the expression levels of four genes, HTR2A, PLXNA2, SRR and TCF4, were
significantly associated with cis SNPs in at least one brain region tested. One gene, SRR, was also involved in a coexpression module that we found to be associated with disease status. In
addition, a substantial number of cis eQTL genes were also involved in the module, suggesting eQTL analysis of brain tissue may identify more reliable susceptibility genes for schizophrenia
than case–control genetic association analyses. In an attempt to facilitate the identification of genetic variations that may underlie the etiology of major psychiatric disorders, we have
integrated the brain eQTL results into a public and online database, Stanley Neuropathology Consortium Integrative Database (SNCID; http://sncid.stanleyresearch.org).
Schizophrenia, bipolar disorder and severe depression are common and highly disabling brain diseases caused by an interaction of genetic and environmental factors.1, 2 However, despite
enormous efforts, the genetic variations that contribute to these diseases and their environmental risk factors remain elusive. Genome-wide association studies have frequently been employed
to identify susceptibility genes and single-nucleotide polymorphisms (SNPs) that may be associated with these mental disorders.3, 4, 5 A number of candidate genes for the disorders have been
reported. For instance, a web resource for schizophrenia, the Schizophrenia Forum (SZgene) database (http://www.szgene.org/), includes results from 1727 genetic association studies and
reports 1008 candidate genes and 8788 polymorphisms in the update on 15 April 2011.6 Despite the numerous candidate genes reported for schizophrenia, the effect size of each variant is small
or moderate and most associated SNPs have failed to be replicated. The need for independent and systematic validation to prioritize further examination of possible candidate genes for
mental disease is widely acknowledged.
Identification of DNA sequence variants that regulate gene expression levels in a relevant tissue is one of the most promising approaches used to initially scan for candidate genes as well
as to prioritize previously identified candidate genes that are associated with complex disease such as psychiatric disorders.7, 8, 9 The identification of a cis association of a SNP with
gene expression levels has been previously used to validate candidate genes for complex traits mapped to the same chromosomal locations.10 Our recent study using an integrative approach that
combined results from genome-wide SNP scans for the cytoarchitectural traits and cis expression quantitative trait loci (eQTL) analysis in the brain tissue revealed two novel candidate
genes associated with cellular abnormalities in the prefrontal cortex of major psychiatric disorders.11 Limited availability of human post-mortem brain tissues is a major obstacle to
obtaining detailed brain expression complex trait loci (eQTL) mapping. Utilization of publicly available resources is an effective alternative strategy that may overcome such limitation. The
Stanley Neuropathology Consortium Integrative Database (SNCID; http://sncid.stanleyresearch.org) is a publicly available and web-based tool that integrates expression microarray data sets
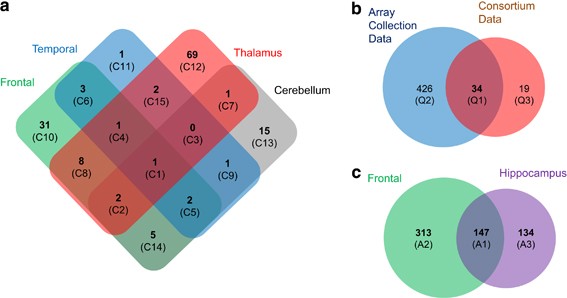
from five brain regions including frontal cortex, temporal cortex, thalamus, cerebellum and hippocampus and genome-wide SNP genotype data sets of subjects in the Stanley Neuropathology
Consortium (SNC) and the Array Collection (AC).12 A total of 1749 neuropathology data sets using the SNC are integrated into the database, which thereby enables one to further explore the
correlations between gene expression levels and quantitative measures of neuropathological markers in the various brain regions. The specific aims of this study are twofold. First, we
explore the candidate genes that may be functionally relevant for major psychiatric disorders by identifying cis associations between SNPs and gene expression in various brain tissues.
Second, we examine the possible functional role of schizophrenia candidate genes that were previously identified in genetic association studies. Thus, we explored cis eQTLs in the four brain
regions, frontal cortex, temporal cortex, thalamus and cerebellum, of SNC subjects and in hippocampus of AC subjects. We also repeated the analysis in frontal cortex data from the AC as a
replication study to examine the overall consensus of cis eQTLs between the two frontal data sets. We then examined whether the expression levels of any candidate genes from the SZgene
database meta-analysis (http://www.szgene.org/) were regulated by cis expressed SNPs (eSNPs) in brain tissues, in order to determine if there were any functional effects on gene expression
of the previously identified schizophrenia susceptibility genes. Finally, we performed a coexpression network analysis between the genes in the frontal cortex that were differentially expre
ssed between schizophrenia and normal controls and the cis eQTL genes in an attempt to identify the potential role of these genes in a disease-specific coexpression module.
Gene expression microarray data from frontal cortex,13 cerebellum, thalamus and temporal cortex14were generated by multiple independent groups using samples from the SNC (N=60), which
contains 15 well-matched cases in each of four groups: schizophrenia, bipolar disorder, major depression and unaffected controls.15 Other sets of microarray data from frontal cortex16, 17and
hippocampus were generated using samples from the AC (N=105). The AC is an independent tissue collection containing 35 cases in each of three groups: schizophrenia, bipolar disorder and
unaffected controls. The groups from both tissue collections are matched for descriptive variables such as age, gender, race, post-mortem interval, mRNA quality, brain pH and hemisphere.
Outlier chip data were excluded in this analysis based on previous quality-control analyses for chip-level parameters such as scaling factor, gene call and average correlation.18 Information
for the microarray studies such as tissue collection, brain region and number of outlier chips is listed in the Supplementary Table S1 online. The confounding effects on the Frozen Robust
Multiarray Analysis (fRMA)-normalized microarray gene expression data were identified using Surrogate Variable Analysis (SVA).19 To adjust disease effect on the gene expression data, we
randomly assign 0 or 1 for the primary variable in the SVA. All covariates from SVA were used in the linear regression to adjust the confounding effects on the gene expression data. The
standardized residuals from the linear regression were used to evaluate the effectiveness of this method on removing confounding variables on two microarray data sets from both the SNC and
AC. Transcripts correlated with potential confounding variables were identified using nonparametric analysis. The continuous variables such as age, brain pH, post-mortem interval and
lifetime exposure to antipsychotics were examined by correlation analysis using R (open source program from Comprehensive R Archive Network (CRAN)). Two categorical variables such as
microarray batch and sex were tested using variance analysis. Adjusted P-values, based on the Hochberg method that were